Introduction
Notre mission est de fournir des soins complets aux patients atteints de maladies néoplasiques du pancréas. Le programme chirurgical offre une expertise de plus de 20 ans dans les tumeurs pancréatiques et comprend des chirurgiens certifiés et spécialisés dans la chirurgie du pancréas et du foie. Les cas sont analysés chaque semaine ou chaque jour en fonction de leur complexité, de manière à combiner les avantages de la multidisciplinarité et la nécessité d'un traitement rapide des tumeurs du pancréas. Cette collaboration nous permet d'apporter une contribution continue qui nous aide à fournir les meilleurs soins possibles à nos patients.
Les consultations
Nos chirurgiens acceptent de nouveaux patients tous les jours. Une clinique générale de consultation est également disponible pour les visites postopératoires et le suivi. Après avoir examiné votre cas, votre chirurgien peut demander une série d'examens, en particulier lorsque le diagnostic n'est pas clair. Les examens radiologiques et diagnostiques, tels que la tomographie assistée par ordinateur, l'imagerie par résonance magnétique, l'échographie endoscopique avec biopsie guidée par ultrasons, sont effectués par l'équipe multidisciplinaire du CHL dans le meilleur délai dans l’objectif de garantir une prise en charge personnalisée et rapide.
La chirurgie des tumeurs pancréatiques et peri-pancréatique
Une chirurgie avec intention curative peut être pratiquée chez les patients présentant des tumeurs pancréatiques, du duodénum, des voies biliaires et de l'ampoule de Vater malignes, que selon des critères bien établi sont considéré opérable soit immédiatement soit après un traitement préopératoire par chimiothérapie. La chirurgie implique une résection pancréatique partielle formelle (pancréaticoduodénectomie/pancréatectomie distale avec splénectomie) ou une pancréatectomie totale. Une lymphadénectomie est également pratiquée, une résection vasculaire peut être effectuée si nécessaire. Une chirurgie épargnant le parenchyme (pancréatectomie moyenne, énucléation) peut être proposée pour les néoplasmes borderline.
La chirurgie pancréatique Mini-Invasive
La chirurgie mini-invasive est pratiquée par de petites incisions dans la paroi abdominale. En chirurgie laparoscopique, les opérations sont effectuées à l'aide d'une caméra vidéo spécialement conçue, qui affiche les images sur des écrans de télévision, et d'instruments spécialement conçus. Au CHL on dispose des plus modernes technologies vidéo 4K ultra HD et 3D qui offrent une visibilité exceptionnelle lors de la chirurgie mini-invasive. Le chirurgien est donc en mesure d'identifier des détails qui ne pourraient pas être affichés avec des technologies vidéo de niveau inférieur. La sécurité des patients est donc considérablement accrue. La chirurgie mini-invasive peut également être réalisée par robotique. Les robots qui pratiquent la chirurgie sont pilotés par des chirurgiens qui dirigent l'opération depuis une console d'ordinateur située dans la salle opératoire à côté du malade. Nos chirurgiens vous indiqueront si votre opération peut être effectuée par laparoscopie ou par robot.
Les traitements palliatifs
Les traitements palliatifs permettent de résoudre les symptômes causés par le cancer du pancréas. Ils peuvent être endoscopiques (par exemple, la pose d'un stent biliaire), percutanés (la pose d'un drainage biliaire transhépatique) ou chirurgicaux (dérivation).
L'hospitalisation et le séjour à l'hôpital
Après avoir effectué tous les examens nécessaires à l'intervention au cours de l'intervention de pré-récupération (ou pendant le séjour dans le service de gastro-entérologie) et une fois l'indication à l'intervention (indication partagée au sein du groupe multidisciplinaire hépato-bilio-pancréatique), le patient est inscrit au programme pour l'intervention proposée.
La date de l'intervention est gérée par le bureau de programmation et une équipe d'informaticiens qui se consacrent à l'optimisation de l'utilisation des lits et des salles d'opération disponibles. Le temps d'attente moyen entre l'évaluation initiale et l'intervention est de 15 jours, sauf s'il est décidé de reporter l'intervention pour optimiser l'état clinique du patient (nutritionnel, par le biais d'un soutien parentéral ou général, par un programme intensif de réadaptation préopératoire). La date de l'intervention est généralement communiquée au patient bien à l'avance afin qu'il puisse prendre ses dispositions.
L'admission se fait généralement la veille de l'opération dans le service de chirurgie générale UF30 (3e étage). Une fois hospitalisé, le patient sera accueilli par l'infirmière qui lui est dédiée et la pré-récupération sera examinée à nouveau par le médecin de service, qui vérifiera la justesse et la présence d'un consentement éclairé à l'intervention proposée. D'autres échantillons de sang seront normalement prélevés pour la demande de produits sanguins qui resteront disponibles pour l'intervention.
Une douche sera prise dans la soirée.
Les opérations de chirurgie du pancréas sont généralement programmées en début de journée. Le jour de l'opération, le patient sera donc conduit à la salle opératoire à 7h30.
Les proches pourront décider de l'opportunité d'un entretien téléphonique après l'opération en donnant au personnel soignant un numéro de téléphone de contact.
Le patient sera conduit au bloc opératoire, où l'anesthésiste qui lui est dédié examinera l'évaluation effectuée pendant la phase préopératoire et, ensuite, pratiquera l'anesthésie. Il est possible que, si le patient a donné son consentement, un petit cathéter péridural soit placé avant d'entrer dans la salle d'opération à des fins analgésiques.
La majorité des patients qui subissent une chirurgie du pancréas sont transférés à l'unité de soins intensifs (3e étage), où ils sont lentement réveillés et extubés.
Au réveil, le patient se retrouvera dans un environnement très technologique, entouré de moniteurs et de pompes qui soutiennent ses fonctions vitales, mais il sera également accompagné d'une infirmière qui lui sera entièrement dédiée et de deux médecins par service, responsables de son rétablissement. En général, il ne sera pas capable de se nourrir lui-même, il trouvera donc un petit tube inséré dans son nez pour garder son estomac dégagé ; une sonde vésicale permettra de mesurer correctement la quantité d'urine produite. Sur l'abdomen, en plus du pansement chirurgical, il y aura un ou deux sacs de drainage pour la collecte des sécrétions abdominales postopératoires.
Généralement, les patients sont peu affectés des douleurs post-chirurgicales : grâce à un cathéter péridurale ou à des perfusions continues d'analgésiques, le contrôle de la douleur est presque toujours bon.
Une fois stable, le patient retournera dans le service, dans sa chambre.
Le programme de récupération postopératoire est assez standardisé et repose sur la mobilisation précoce du patient, le contrôle de la douleur, la surveillance des éventuelles complications, avec des prélèvements sanguins et des contrôles cliniques et de laboratoire sur le liquide de drainage abdominal.
Si tout se passe bien, la sonde dans l'estomac est retirée au bout de quelques jours et le patient commence à boire, en augmentant progressivement l'alimentation par la bouche au cours des jours suivants. La sonde vésicale est également retirée vers le troisième jour, les drains, selon les déterminations du laboratoire, sont d'abord partiellement retirés puis, après quelques jours, complètement enlevés.
Au bout d'une semaine environ, le patient est généralement capable de se nourrir lui-même, mais pas suffisamment pour couvrir ses besoins nutritionnels (en fait, le soutien calorique par voie intraveineuse est maintenu). Cependant, il est capable, si on l'aide, de se déplacer dans la pièce. L'autonomie fonctionnelle du patient, surtout dans les premiers jours, est bien meilleure si l'intervention est réalisée par laparoscopie ou robotique.
En général, entre le septième et le dixième jour, une tomographie assistée par ordinateur est effectuée pour exclure la présence d'épanchements abdominaux et vérifier l'intégrité des structures vasculaires.
Le patient peut donc rentrer à domicile ou parfois envoyé dans des établissements de réadaptation protégés.
Le retour au domicile
Le retour à la maison représente toujours pour le patient un moment de grande joie et en même temps d'inquiétude : nous voulons nous assurer qu'en tout état de cause, les patients en voie de guérison ne sont jamais laissés seuls : il est toujours possible de contacter un médecin du service en appelant les numéros de téléphone indiqués au moment de la sortie. Le patient lui-même sera toujours examiné à plusieurs reprises en consultation.
Les difficultés que les patients rencontrent sont habituellement liées au régime alimentaire à suivre, à la gestion de la fatigue qu'ils ressentent, ou à la gestion de la stéatorrhée, de la diarrhée ou du diabète, il nous appartiendra de répondre à ces questions et d'orienter les patients vers d'autres spécialistes (ex : diabétologue, diététicien ...) en cas de besoin.
Le pancréas est un organe situé au plus profond de l'abdomen.
Il mesure environ 15 à 20 centimètres de long et pèse approximativement 70 à 160 grammes. Le pancréas est anatomiquement divisé en quatre parties :
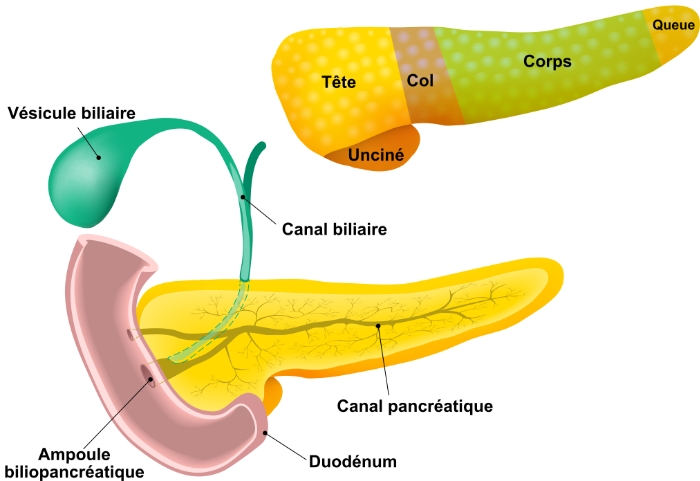
- La tête. La tête est la partie la plus large du pancréas, et elle est logée dans la courbe du duodénum. Le processus d'incontinence est un prolongement de la tête du pancréas (bien qu'ayant une genèse embryologique différente) qui englobe les vaisseaux mésentériques supérieurs.
- Le col. Le col jaillit de la partie supérieure droite de l'avant de la tête. Il mesure environ 2,5 cm de long. Sa surface postérieure est en relation avec la veine porte.
- Le corps. La partie centrale est appelée le corps. Il a une forme quelque peu prismatique et est recouvert par la surface postérieure de l'estomac qui repose sur lui.
- Queue. La partie la plus fine est appelée queue. Elle s'étend vers la gauche, et son extrémité est en contact avec la rate.
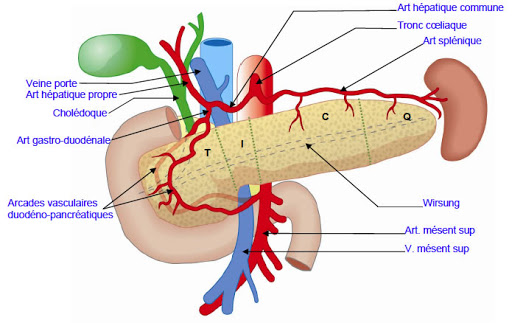
Le pancréas est en relation avec de nombreux vaisseaux sanguins importants. La veine cave inférieure et l'aorte sont situées à l'arrière de la tête/col du pancréas. Le trépied cœliaque part de l'aorte près du bord supérieur du corps pancréatique. Il se ramifie dans l'artère gastrique gauche, l'artère hépatique commune et l'artère splénique. L'artère mésentérique supérieure alimente l'intestin grêle. Elle prend naissance dans l'aorte à quelques centimètres sous le tronc cœliaque, est fixée à la surface postérieure de la tête pancréatique et descend à travers le processus d'incontinence. La veine mésentérique supérieure remonte sur le côté droit de l'artère et, derrière le cou, se joint à la veine splénique pour former la veine porte. La veine portale draine le sang du tractus gastro-intestinal et de la rate vers le foie.
Le pancréas est une glande, ayant à la fois des fonctions exocrines et endocrines.
Le composant exocrine du pancréas (acini pancréatique) produit des sucs pancréatiques. Ces jus contiennent de l'eau, des ions et des enzymes qui aident à décomposer les aliments. Les sucs circulent dans un système de conduits (appelés conduits secondaires) menant au conduit pancréatique principal (ou conduit de Wirsung), qui s'étend transversalement de gauche à droite à travers la substance du pancréas. Les sucs pancréatiques s'écoulent par le canal principal (canal de Wirsung) jusqu'au duodénum, la première partie de l'intestin grêle. À l'intérieur de la paroi duodénale, le canal pancréatique principal se termine par un orifice commun à celui-ci et au canal biliaire commun appelé ampoule de Vater. La zone entourant l'ampoule de Vater est appelée région périampullaire. Un canal supplémentaire, appelé canal pancréatique accessoire ou canal de Santorin, est émis par le canal pancréatique dans le col du pancréas et débouche dans le duodénum à environ 2,5 cm au-dessus de la papille duodénale, dans une minuscule structure appelée papille mineure. Elle reçoit les canaux de la partie inférieure de la tête.
Le pancréas est également une glande endocrine qui produit de l'insuline et d'autres hormones. Les régions du pancréas contenant des cellules productrices d'hormones sont appelées îlots de Langerhans. Les hormones produites dans les îlots de Langerhans sont sécrétées directement dans le flux sanguin et voyagent dans tout l'organisme. Elles aident l'organisme à utiliser ou à stocker l'énergie provenant des aliments. Par exemple, l'insuline et le glucagon aident à contrôler la quantité de sucre dans le sang.
Les tumeurs pancréatiques et périampullaires constituent un groupe hétérogène de lésions provenant des acini pancréatiques, des cellules des îlots de Langerhans, des canaux pancréatiques, du canal cholédoque, de l'ampoule de Vater et du duodénum. Leur comportement biologique et le pronostic varient fortement en fonction du type de tumeur et du stade de la tumeur.
La thérapie des tumeurs pancréatiques est principalement chirurgicale.
Les différentes tumeurs pancréatiques:
- L’adénocarcinome pancréatique
- Le cholangiocarcinome distale (tumeur de la voie biliaire)
- La Tumeur Duodénale
- Le GIST duodénale
- Les Tumeur de la région Ampullaire
- Les Tumeur Kystiques du pancréas
- Les Tumeurs Endocrines du pancréas
1. L’adénocarcinome pancréatique
Généralités
L'adénocarcinome canalaire est de loin le type de néoplasmes pancréatiques le plus courant. C'est une maladie très agressive, qui représente la quatrième cause de décès par cancer dans le monde occidental. Le risque de développer un adénocarcinome canalaire pancréatique augmente avec l'âge (l'âge médian au moment du diagnostic est de 71 ans), et les hommes ont 30 % plus de chances de développer ce néoplasme que les femmes. La grande majorité des adénocarcinomes canalaires est sporadique, seule une petite fraction étant familiale. L'adénocarcinome canalaire débute lorsque, en raison d'une lésion de l'ADN, les cellules du pancréas commencent à se développer de manière incontrôlée et forment une tumeur. Les bases génétiques et moléculaires de l'adénocarcinome canalaire du pancréas sont encore mal comprises et font l'objet de recherches intensives.
Les facteurs de risque connus de l'adénocarcinome canalaire pancréatique sont les suivants:
- L'âge (> 60 ans)
- Le sexe masculin
- Fumer des cigarettes
- L'abus d'alcool
- Obésité et diabète mellitus
- Pancréatite chronique
- Autres néoplasmes du pancréas (néoplasmes mucineux papillaires intraconduits)
- Antécédents familiaux de cancer du pancréas
- Syndromes génétiques tels que la mutation BRCA2 et la polypose colique
Malheureusement, très peu d'adénocarcinomes canalaires pancréatiques sont découverts précocement. Les patients présentent généralement des symptômes peu spécifiques ou même aucun symptôme jusqu'à ce que le cancer se soit propagé à d'autres organes. Les examens radiologiques de routine (tels que les ultrasons) ou les examens physiques ne permettent pas de détecter les petites tumeurs. À l'heure actuelle, il n'existe pas de tests sanguins permettant de détecter les cancers précoces du pancréas. Les symptômes gastro-intestinaux les plus courants associés à l'adénocarcinome canalaire du pancréas sont :
- Faiblesse
- Problèmes digestifs (manque d'appétit, dyspepsie ; selles pâles, volumineuses et graisseuses)
- Douleurs épigastriques et dorsales
- Perte de poids
- Apparition soudaine du diabète sucré
Les cancers qui commencent dans la tête du pancréas provoquent généralement des symptômes alors qu'ils sont encore assez petits. Il s'agit notamment de la jaunisse et de l'obstruction duodénale, qui peuvent permettre de retrouver ces tumeurs à un stade plus précoce.
- La jaunisse : il s'agit du jaunissement des yeux et de la peau causé par l'accumulation de bilirubine dans le corps. La bilirubine est une substance jaune-brun foncé qui est fabriquée dans le foie. Normalement, le foie excrète la bilirubine dans la bile, qui s'écoule dans les intestins par le canal biliaire commun. Lorsque la portion de bile commune passant dans la tête du pancréas est comprimée ou infiltrée par la tumeur, la bile ne peut pas atteindre les intestins, et le niveau de bilirubine s'accumule. Le premier signe de la jaunisse est l'obscurcissement de l'urine, qui devient brune. En outre, une personne peut remarquer que ses selles deviennent plus claires. Lorsque la bilirubine s'accumule dans la peau, elle devient jaune et commence à démanger. Il est à noter que l'adénocarcinome canalaire du pancréas et d'autres tumeurs survenant dans la zone périampullaire, notamment le cancer ampullaire et le cholangiocarcinome distal, ne sont pas la cause la plus fréquente de la jaunisse. D'autres causes, telles que les calculs biliaires, l'hépatite et d'autres maladies du foie, sont beaucoup plus fréquentes.
- Obstruction intestinale : le néoplasme peut s'infiltrer à l'extrémité de l'estomac ou du duodénum, les bloquant en partie. Cela peut provoquer des nausées, des vomissements et des douleurs qui ont tendance à s'aggraver après un repas.
Les cancers qui commencent dans le corps ou la queue du pancréas ne compriment pas le canal biliaire avant de s'être propagés dans le pancréas. À ce moment-là, le cancer peut également s'être propagé au-delà du pancréas.
Lorsqu'un adénocarcinome canalaire pancréatique est suspecté par un médecin, il est fortement conseillé de l'adresser à une équipe multidisciplinaire ayant de l'expérience dans ce domaine. L'estimation de l'étendue de la maladie sur l'imagerie transversale (classification de la tumeur) est le facteur le plus important dans le choix des options de traitement et la prévision des perspectives du patient. Une biopsie peut également être pratiquée pour s'assurer du diagnostic. Pour plus d'informations sur le diagnostic et la classification de l'adénocarcinome canalaire pancréatique.
Le bilan diagnostique et la classification
La modalité d'imagerie la plus répandue et la plus disponible est l'échographie transabdominale, qui permet d'identifier les masses pancréatiques. Cependant, l'échographie est peu précise dans la représentation des caractéristiques de la tumeur, car le pancréas est situé au plus profond de l'abdomen. À l'échographie, l'adénocarcinome canalaire pancréatique apparaît comme une masse hypoéchogène (plus sombre que le pancréas environnant). La dilatation associée du canal cholédoque, lorsqu'elle est présente, est bien détectable.
Chez les patients atteints de jaunisse, il peut être nécessaire de placer un stent (un petit tube en plastique ou en métal) pour soulager un canal cholédoque obstrué et résoudre la jaunisse. Cela se fait généralement par une procédure endoscopique appelée CPRE (cholangiopancréatographie rétrograde endoscopique). La procédure comprend deux phases distinctes :
Phase diagnostique : un très petit cathéter est guidé par endoscopie à travers l'ampoule de Vater dans le canal biliaire commun. Une petite quantité d'agent de contraste est ensuite injectée, et des radiographies sont prises. Ce colorant délimite le canal biliaire et le canal pancréatique, et montre le site et la morphologie de l'obstruction biliaire qui pourrait être due à un cancer du pancréas.
Phase opératoire : une petite incision est pratiquée sur l'orifice ampullaire (papillotomie), puis le stent est passé à travers l'endoscope et est placé dans le canal biliaire. Le stent aide à maintenir le canal cholédoque ouvert et résiste à la compression du cancer environnant.
Au cours d'une CPRE, il est possible d'effectuer une biopsie par brossage du canal cholédoque pour vérifier la présence de cellules néoplasiques, en particulier si le canal pancréatique a été largement infiltré par les néoplasmes pancréatiques. Ce type de biopsie est cependant peu précis. La CPRE est une procédure invasive qui est associée à un profil de complication spécifique, notamment une pancréatite aiguë sévère. De plus, la canulation ampullaire peut être difficile, et la mise en place d'un fil-guide et d'un stent peut échouer si le canal biliaire est petit ou tortueux. Dans de tels cas, la jaunisse peut être résolue en plaçant un drainage biliaire transhépatique percutané (PTBD). Un mince cathéter est placé par voie percutanée dans les voies biliaires intrahépatiques jusqu'à la voie biliaire commune.
La découverte d'une masse pancréatique suspecte d'adénocarcinome canalaire nécessite une imagerie en coupe transversale de haute qualité pour confirmer le diagnostic radiologique et mettre en évidence le néoplasme. Un système de classification est un moyen standardisé par lequel l'équipe de soins du cancer décrit l'étendue de la propagation d'un cancer et contient différents éléments d'information, notamment
- La taille de la tumeur primaire
- si la tumeur s'est propagée aux organes ou vaisseaux voisins
- si la tumeur s'est propagée aux ganglions lymphatiques voisins
- si la tumeur s'est propagée (métastasée) à des organes éloignés
Les tests d'imagerie permettant de diagnostiquer et de mettre en évidence un adénocarcinome canalaire pancréatique:
- Le scanner abdominal et pulmonaire avec produit de contraste (TAP).
Le TAP est un test à rayons X (il utilise des radiations ionisantes) qui produit des images détaillées en coupe transversale du corps. Des logiciels de reconstruction 3D permettent de caractériser la tumeur de manière détaillée. Les scans CT montrent le pancréas assez clairement et peuvent souvent confirmer la localisation de la tumeur, qui semble hypodense et mal mise en valeur. Les scanners peuvent également montrer les organes proches du pancréas, ainsi que les ganglions lymphatiques et les organes éloignés où le cancer pourrait s'être propagé.
- L'imagerie par résonance magnétique (IRM).
L'IRM utilise des ondes radio et des aimants puissants au lieu de rayons X. Il s'agit d'une modalité d'imagerie multiplanaire. L'IRM est moins utilisée que le scanner pour la stadification de l'adénocarcinome canalaire pancréatique, mais elle peut être très utile lorsque la tumeur présente une morphologie complexe (lésion mixte avec des zones solides et kystiques), ou lorsque le diagnostic différentiel avec d'autres néoplasmes pancréatiques n'est pas clair.
- L'échographie avec renforcement du contraste (CEUS).
Il s'agit d'une nouvelle technique d'imagerie qui implique l'utilisation d'agents de contraste à microbulles pour montrer des informations en temps réel sur la perfusion des tissus. Lors de l'examen CEUS, l'adénocarcinome canalaire montre généralement une faible amélioration pendant toutes les phases dynamiques. La classification loco-régionale par CEUS de l'adénocarcinome canalaire entre des mains expertes est précise. Les marges et la taille de la lésion sont plus visibles, ce qui améliore la détection des infiltrations vasculaires.
- L'échographie écoendoscopique (EUS).
L'échographie endoscopique est réalisée à l'aide d'une sonde à ultrasons qui est fixée à l'extrémité d'un endoscope. Cela permet une vision directe du duodénum et de la région papillaire ainsi qu'une échographie très détaillée du pancréas, qui se trouve à côté du duodénum. C'est probablement mieux qu'un scanner pour repérer les petites tumeurs. Si une tumeur est détectée, une biopsie trans-gastrique ou trans-duodénale peut être effectuée au cours de cette procédure.
- La tomographie par émission de positrons (TEP).
La TEP implique l'utilisation d'une très faible dose d'un radiotraceur intraveineux (connu sous le nom de 18-fluorodésoxyglucose ou FDG). Les cellules néoplasiques absorbent une grande quantité de FDG, qui est détectée par le scanner. Ces images fonctionnelles ont cependant une résolution spatiale insuffisante ; c'est pourquoi la TEP-scan est combinée à la tomodensitométrie (TEP-CT) pour fournir des images détaillées de la tumeur primaire. La TEP-TDM peut être particulièrement utile pour repérer un cancer qui s'est propagé au-delà du pancréas.
Dans le cadre du diagnostic de l'adénocarcinome canalaire du pancréas, on mesure souvent des taux sériques d'environ 19-9. Le Ca 19-9 est un simple test sanguin qui mesure le niveau d'antigènes libérés par les cellules tumorales du pancréas. Les taux de Ca 19,9 sont élevés dans le sang de nombreux patients atteints d'adénocarcinome canalaire pancréatique. Cependant, il existe aussi des maladies non cancéreuses qui provoquent un taux élevé de Ca 19,9, comme les calculs biliaires, la pancréatite, la mucoviscidose, les maladies du foie, les maladies pulmonaires et thyroïdiennes. De plus, le taux de Ca 19-,9 peut être élevé chez les personnes souffrant d'une obstruction des voies biliaires, ce qui est le cas de nombreux patients atteints d'adénocarcinome canalaire pancréatique. Au contraire, chez les patients qui n'ont pas l'antigène Lewis (une protéine du groupe sanguin sur les globules rouges), ce qui représente environ 10% de la population caucasienne, le Ca19-9 n'est pas exprimé, même chez ceux qui ont de grosses tumeurs. C'est pourquoi le Ca 19-,9 n'est pas particulièrement utile comme test de diagnostic du cancer du pancréas. Une fois le diagnostic de cancer du pancréas confirmé et si le taux de Ca 19-9 de l'individu était élevé avant le traitement, le test Ca 19-9 peut être utilisé comme facteur de pronostic.
Une fois la caractérisation radiologique obtenue (taille de la tumeur, relation avec les vaisseaux péripancréatiques, état des ganglions lymphatiques, présence de métastases), ces informations sont combinées pour attribuer un stade. La classification actuelle est basée sur le système TNM (tumeur/ état ganglionnaire/métastase), selon l'American Joint Commitee on Cancer (AJCC, www.cancerstaging.org). Le stade de la tumeur est exprimé en chiffres romains I à IV.
Voici les groupes de stades de l'AJCC des adénocarcinomes canalaires pancréatiques:
- Stade IA : La tumeur est confinée au pancréas et mesure moins de 2 cm. Elle ne s'est pas propagée aux ganglions lymphatiques voisins ou à des sites distants.
- Stade IB : la tumeur est confinée au pancréas et mesure plus de 2 cm. Elle ne s'est pas propagée aux ganglions lymphatiques voisins ou à des sites distants.
- Stade IIA : La tumeur se développe en dehors du pancréas, mais pas dans l'artère mésentérique supérieure ou le tronc cœliaque. Elle ne s'est pas étendue aux ganglions lymphatiques voisins ou à des sites distants.
- Stade IIB : la tumeur est soit confinée au pancréas, soit en croissance en dehors du pancréas, mais pas dans l'artère mésentérique supérieure ou le tronc cœliaque. Elle s'est propagée aux ganglions lymphatiques voisins mais pas à des sites distants.
- Stade III : La tumeur se développe en dehors du pancréas dans l'artère mésentérique supérieure ou le tronc cœliaque. Elle peut ou non s'être propagée aux ganglions lymphatiques voisins. Elle ne s'est pas étendue à des sites distants.
- Stade IV : Le cancer s'est étendu à des sites distants.
La classification radiologique divise l'adénocarcinome canalaire pancréatique en groupes selon qu'il est probable ou non qu'il puisse être enlevé chirurgicalement :
- Tumeur opérable ou potentiellement opérable (stade IA, IB, IIA, IIB)
- Tumeur localement avancée (stade III)
- Tumeur métastatique (stade IV)
La tumeur opérable ou potentiellement opérable
Au moment du diagnostic, seuls 20 % environ des adénocarcinomes canalaires pancréatiques semblent pouvoir faire l'objet d'une résection chirurgicale potentiellement curative.
Comme on l'a vu dans la section sur le bilan diagnostique et la classification, un carcinome canalaire pancréatique est considéré comme opérable lorsqu'il ne s'étend pas à l'artère mésentérique supérieure et/ou au tronc cœliaque, et lorsqu'il ne s'est pas propagé à d'autres organes distants (en particulier le foie et le poumon). L'extension de la tumeur dans la veine mésentérique supérieure/la veine portale n'est plus une contre-indication à la chirurgie radicale, mais les résections pancréatiques avec résection veineuse synchrone sont des opérations très complexes associées à une morbidité accrue.
Bien que les récents progrès de l'imagerie en coupe transversale permettent une évaluation détaillée du pancréas, la précision de la classification radiologique n'est pas de 100%. Dans certains cas, il peut même être difficile de déterminer avec précision le stade du cancer du pancréas à l'aide de tests d'imagerie. Par conséquent, lorsqu'il y a de bonnes chances que la tumeur puisse être complètement enlevée, une opération est entreprise, et l'exploration chirurgicale joue toujours le rôle clé pour l'évaluation finale de l’opérabilité. Parfois, on découvre une maladie localement avancée ou métastatique inattendue. Dans ce cas, le chirurgien peut poursuivre l'opération en tant que procédure palliative pour soulager ou prévenir les symptômes.
Toutefois, l'anatomie de la tumeur n'est pas le seul élément à prendre en compte. D'autres facteurs liés au patient sont tout aussi importants pour le processus de décision. Parmi les plus importants, citons les comorbidités et l'état fonctionnel. Les comorbidités font référence à d'autres maladies dont le patient peut souffrir, telles que les maladies cardiaques ou le diabète. L'état fonctionnel fait référence à l'état nutritionnel, à la capacité de subir une intervention chirurgicale majeure et de fonctionner de manière autonome après l'opération. L'évaluation de l’opérabilité nécessite donc une évaluation complexe de l'anatomie de la tumeur, de l'âge, des comorbidités, de l'état fonctionnel et des résultats d'une analyse sanguine (Ca 19.9) pour déterminer le profil risque-bénéfice de l'opération.
Dans certains cas, un traitement néoadjuvant peut être conseillé. La thérapie néoadjuvante est une thérapie médicale (chimio ou chimio-radiothérapie) administrée dans les cas de maladies opérable ou à la limite de l’opérabilité avant l'opération. La justification de la thérapie néoadjuvante dans le cas du cancer du pancréas est multiple. La thérapie préopératoire peut théoriquement stériliser l'étendue périphérique de l'infiltration tumorale, diminuer le volume de la tumeur et la maladie nodale régionale. En outre, les patients qui reçoivent un traitement néoadjuvant ont plus de chances de terminer leur traitement complet que les patients qui reçoivent une chimiothérapie postopératoire. En outre, la thérapie néoadjuvante administrée à des tissus non disséqués et bien oxygénés peut maximiser tout bénéfice cytotoxique tiré du traitement. Enfin, et c'est peut-être le plus important, les patients qui présentent une progression de la maladie au cours de leur traitement néoadjuvant se considèrent comme de mauvais répondants qui ont moins de chances de tirer profit de la résection et peuvent renoncer à la morbidité de la résection pancréatique. Les résultats des études évaluant le rôle de la thérapie néoadjuvante sont prometteurs, malgré l'absence de grands essais randomisés.
Les résections chirurgicales standard pour l'adénocarcinome canalaire pancréatique comprennent la pancréaticoduodénectomie et la pancréatectomie gauche avec splénectomie. L'objectif de la résection chirurgicale est d'éliminer complètement la tumeur, sans laisser de résidu de cancer. Ce concept est connu sous le nom de chirurgie R0 (sans maladie résiduelle). En outre, les résections pancréatiques pour le cancer comprennent la lymphadénectomie régionale. Le rôle de la dissection étendue des ganglions lymphatiques est controversé et ne semble pas être bénéfique. Après la résection, une rection congelée peropératoire de la marge de résection pancréatique est effectuée pour éliminer la maladie microscopique résiduelle (R1) dans le reste du pancréas. Une extension de la résection, jusqu'à une pancréatectomie totale avec splénectomie, peut être nécessaire lorsque la marge de résection est positive pour les cellules tumorales.
L'échantillon de résection est examiné par le pathologiste. L'examen histologique confirme le diagnostic et attribue le stade pathologique de la tumeur (selon le système TNM, AJCC, www.cancerstaging.org). Bien qu'elles ne fassent pas officiellement partie du système TNM, d'autres caractéristiques histologiques sont d'une importance primordiale. Le grade du cancer (l'aspect anormal des cellules sous le microscope) est indiqué sur une échelle allant de G1 à G4, les cancers G1 ressemblant le plus aux cellules normales et offrant les meilleures perspectives. Comme on l'a vu, un autre facteur important est la clairance de la tumeur, que celle-ci soit entièrement enlevée ou non. Ce facteur est indiqué sur une échelle allant de R0 (où toute la tumeur visible et microscopique a été enlevée) à R2 (où une partie de la tumeur visible n'a pas pu être enlevée). En outre, les ganglions lymphatiques sont analysés pour rechercher une implication du cancer (statut des ganglions lymphatiques). Le rapport entre le nombre de ganglions lymphatiques positifs et le nombre total de ganglions lymphatiques prélevés (rapport des ganglions lymphatiques) s'est avéré pertinent pour le pronostic.
Les patients reçoivent une chimiothérapie ou une chimio-radiothérapie après l'ablation chirurgicale du cancer pour tenter d'éliminer les cellules cancéreuses laissées sur place. Ce type de traitement est appelé traitement adjuvant et réduit le risque de récidive du cancer.
Les patients qui ont subi une résection pancréatique potentiellement curative seront inscrits à un suivi périodique strict, consistant en un examen clinique détaillé, une imagerie transversale et une mesure du Ca 19,9 sérique.
La tumeur localement avancé
L'adénocarcinome canalaire pancréatique est défini comme localement avancé (stade III) lorsqu'il s'est étendu à l'artère mésentérique supérieure et/ou au tronc cœliaque. L'adénocarcinome de la tête du pancréas s'étend généralement à l'artère mésentérique supérieure, qui alimente l'intestin en sang riche en oxygène ; les tumeurs de la queue du pancréas s'étendent plus souvent au tronc cœliaque, dont les branches alimentent le foie, l'estomac et la rate. Lorsque l'imagerie en coupe transversale révèle un adénocarcinome canalaire pancréatique localement avancé, il est nécessaire d'obtenir un diagnostic pathologique (généralement cytologique). La procédure la plus souvent utilisée pour diagnostiquer un adénocarcinome canalaire pancréatique est appelée biopsie par aspiration à l'aiguille fine. Pour ce test, une fine aiguille est insérée à travers la peau et dans le pancréas. Une échographie est utilisée pour examiner la position de l'aiguille et s'assurer qu'elle est bien dans la tumeur. Les biopsies peuvent également être effectuées à l'aide d'ultrasons endoscopiques, en plaçant l'aiguille directement à travers la paroi de l'estomac ou à travers le duodénum dans la tumeur. Dans les deux cas, de petits échantillons de tissu peuvent être prélevés à l'aide de l'aiguille. Les échantillons de tissus sont ensuite examinés au microscope. Les cellules anormales sont trouvées et examinées par le pathologiste, qui décide d'un diagnostic.
Si le néoplasme a provoqué des symptômes, un traitement palliatif peut être nécessaire pour les soulager :
Un stent en plastique ou en métal peut être placé par voie endoscopique pour soulager la jaunisse causée par l'obstruction du canal cholédoque. Ce traitement est très courant dans les néoplasmes de la tête du pancréas. Une autre solution consiste à mettre en place un drainage biliaire percutané. Pour plus d'informations, cliquez ici.
Si nécessaire, la chirurgie peut rediriger le flux de bile du canal cholédoque directement dans l'intestin grêle, en contournant le pancréas (derivation). La connexion de l'estomac au duodénum peut également être redirigée à ce moment-là pour soulager ou prévenir l'obstruction du duodénum. En outre, des biopsies peropératoires peuvent être effectuées.
Le traitement de première ligne de l'adénocarcinome canalaire pancréatique localement avancé est la thérapie médicale. La chimiothérapie implique l'utilisation de médicaments pour tuer les cellules cancéreuses, et peut être administrée par voie intraveineuse ou orale. Ces médicaments sont généralement administrés par cycles, avec une alternance de périodes de traitement et de récupération, et peuvent être administrés seuls ou en conjonction avec la radiothérapie (chimio-radiothérapie). L'équipe d'oncologie choisit le meilleur plan de traitement pour chaque patient. Une fois le programme de traitement terminé, on procède à une nouvelle répartition des soins. Cela implique un examen clinique détaillé, la mesure du taux de Ca 19,9 dans le sérum et l'imagerie en coupe (CT-scan ou PET-CT). Les résultats sont discutés au sein de notre équipe multidisciplinaire. Les résultats obtenus après un traitement médical de première ligne pour un adénocarcinome canalaire pancréatique localement avancé comprennent :
La régression de la maladie (down-staging). Cela signifie que la maladie est devenue opérable,, au moins sur la base de l'imagerie. Le chirurgien peut décider d'une exploration chirurgicale, s'il est probable que le néoplasme puisse être complètement enlevé chirurgicalement. Grâce aux nouveaux schémas de chimiothérapie, on signale de plus en plus souvent des résections pancréatiques pratiquées après une réduction de la maladie.
La maladie est stable. Cela signifie qu'aucune nouvelle tumeur ne s'est développée, et que le néoplasme ne s'est pas propagé à de nouvelles régions du corps (en d'autres termes, le néoplasme ne s'améliore pas ou ne s'aggrave pas). Dans une telle situation, la prise en charge est adaptée à chaque patient, après une discussion multidisciplinaire. Certain patients pourront bénéficier d’un résection pancréatique complexe avec résection vasculaire associé, d’autre d’une chimiothérapie de deuxième ligne ou une chimio-radiothérapie supplémentaire peut être conseillée, ainsi que l'inclusion dans des protocoles expérimentaux.
Progression de la maladie. Cela signifie que la tumeur a progressé localement (a grandi) ou s'est étendue à d'autres organes du corps (s'est métastasée). Le site métastatique le plus courant est le foie. Dans une telle situation, une chimiothérapie de deuxième ligne peut être indiquée, ainsi que l'inclusion dans des essais cliniques expérimentaux. Dans tous les cas, les patients seront suivis et réévalués périodiquement.
La tumeur métastatique
Le cancer métastatique est un cancer qui s'est propagé, par le biais du système sanguin ou lymphatique, de l'endroit où il a débuté à un autre endroit du corps. La capacité d'une cellule cancéreuse à se métastaser dépend de ses propriétés individuelles, des propriétés des cellules non cancéreuses, y compris les cellules du système immunitaire, présentes à l'endroit initial, et des propriétés des cellules qu'elle rencontre dans le système lymphatique ou la circulation sanguine et à la destination finale dans une autre partie du corps. L'adénocarcinome canalaire pancréatique se métastase le plus souvent dans le foie, les ganglions lymphatiques distants, le péritoine et le poumon (stade IV). Le traitement médical standard de l'adénocarcinome canalaire pancréatique métastatique comprend généralement une chimiothérapie. Il semble qu'il existe des combinaisons intéressantes et potentiellement prometteuses de plusieurs agents médicamenteux de traitement médical conventionnel qui sont en pratique et à l'étude pour le traitement de l'adénocarcinome canalaire pancréatique métastatique. En outre, un large éventail d'approches de traitement médical en mode unique est actuellement en cours d'essais cliniques contre l'adénocarcinome du pancréas. Il s'agit notamment de certaines des thérapies expérimentales les plus récentes qui visent davantage les cibles moléculaires et l'interruption des voies de signalisation génétique, de nouveaux médicaments de chimiothérapie et même de vaccins contre le cancer.
Des schémas thérapeutiques individualisés, adaptés par des oncologues experts, impliquant des agents uniques ou une thérapie combinée pour l'adénocarcinome canalaire pancréatique métastatique, peuvent prolonger la survie et la qualité de vie. Enfin, les essais cliniques restent une option.
2. Le cholangiocarcinome distale (tumeur de la voie biliaire)
Généralités
Le système biliaire est un système de conduits en forme d'arbre qui relie le foie et la vésicule biliaire au duodénum.
Le système biliaire permet à la bile, un liquide épais qui est produit dans le foie et stocké dans la vésicule biliaire, de s'écouler dans le duodénum et d'aider à la digestion des graisses. Le système biliaire trouve son origine dans le foie. De petits tubes, semblables à des capillaires et appelés canaux biliaires intrahépatiques, drainent la bile des cellules du foie vers des branches de plus en plus grosses, qui se terminent par un tube appelé canal biliaire commun ou canal biliaire extra-hépatique. Le canal biliaire sort du foie et s'écoule dans le duodénum. La première moitié du canal biliaire commun passe par le ligament hépato-duodénal, ainsi que par l'artère hépatique et la veine porte ; la seconde moitié passe par la tête pancréatique et rejoint le canal pancréatique principal au niveau de l'ampoule de Vater. La vésicule biliaire est un réservoir qui retient la bile jusqu'à ce que les aliments atteignent les intestins. Elle est reliée par un petit conduit, appelé canal cystique, aux voies biliaires communes à environ un tiers de la descente du canal biliaire depuis le foie.
Les adénocarcinomes des voies biliaires (ou cholangiocarcinomes) sont un groupe de néoplasmes agressifs provenant des cellules épithéliales qui tapissent les voies biliaires. Selon leur site primaire, ils peuvent être divisés en deux catégories :
- le cholangiocarcinome intra-hépatique
- le Cholangiocarcinome extra-hépatique (ou du canal cholédoque)
Le cholangiocarcinome extra-hépatique se subdivise encore en trois types :
- Le cholangiocarcinome périhilarien (ou tumeur de Klatskin). Ce néoplasme commence là où les canaux hépatiques gauche et droit (les deux principales branches intra-hépatiques) se rejoignent dans le canal cholédoque commun, au point où il quitte le foie. C'est la partie du système biliaire où les néoplasmes apparaissent généralement (2/3 des cas)
- Cholangiocarcinome du canal biliaire moyen, le type le moins fréquent.
- Cholangiocarcinome distal. Ce type de cancer survient dans les voies intra-pancréatiques ou péri-ampullaires du canal cholédoque commun.
Le cholangiocarcinome primaire est une maladie rare (2500 nouveaux cas sont diagnostiqués chaque année aux États-Unis), son incidence est plus importante en Asie.
Les principaux symptômes du cholangiocarcinome extra-hépatique sont les suivants :
- La jaunisse
- Perte de poids
- Faiblesse
- Problèmes digestifs (perte d'appétit, dyspepsie)
- Douleur dans le quadrant supérieur droit de l'abdomen
Les cancers qui débutent dans le canal cholédoque extra-hépatique provoquent généralement une jaunisse, c'est-à-dire le jaunissement des yeux et de la peau causé par l'accumulation dans l'organisme de bilirubine, une substance contenue dans la bile. Lorsque le canal biliaire commun est comprimé ou infiltré par la tumeur, la bile ne peut pas atteindre les intestins et le niveau de bilirubine s'accumule. Le premier signe de la jaunisse est l'assombrissement de l'urine, qui devient brune. En outre, une personne peut remarquer que ses selles deviennent plus claires. Lorsque la bilirubine s'accumule dans la peau, elle devient jaune et commence à démanger. Il est à noter que le cholangiocarcinome et les autres tumeurs survenant dans la tête du pancréas et dans la zone périampullaire ne sont pas la cause la plus fréquente de la jaunisse. D'autres causes, telles que les calculs biliaires, l'hépatite et d'autres maladies du foie, sont beaucoup plus fréquentes. Les patients souffrant d'un ictère obstructif persistant risquent de développer une cholangite, une infection du canal biliaire. Les symptômes caractéristiques sont la fièvre, les frissons et les douleurs abdominales, et dans les cas graves. La cholangite peut être une affection grave, associée à une septicémie. Lorsqu'un patient présente une cholangite, il est nécessaire de soulager l'obstruction du canal biliaire en plaçant un stent (plastique ou métallique) ou en mettant en place un drainage biliaire transhépatique percutané (DBCT).
Lorsqu'un cholangiocarcinome est suspecté par un médecin, il est fortement conseillé de le référer à une équipe multidisciplinaire ayant une expérience dans ce domaine. L'estimation de l'étendue de la maladie sur l'imagerie (classification de la tumeur) est le facteur le plus important dans le choix des options de traitement et la prévision des perspectives du patient. Une biopsie peut également être pratiquée pour s'assurer du diagnostic. Pour plus d'informations sur le bilan diagnostique et la classification du cholangiocarcinome.
Le bilan diagnostique et la classification
Le symptôme le plus fréquent du cholangiocarcinome, qui conduit à un examen diagnostique, est la jaunisse. La première modalité d'imagerie pour explorer le système biliaire est l'échographie, qui décrit bien la dilatation des voies biliaires intra et extra-hépatiques. Cependant, l'échographie est peu précise dans la description de la morphologie du rétrécissement des voies biliaires et peut ne pas visualiser le néoplasme primaire. En fait, le cholangiocarcinome extra-hépatique peut ne pas former une masse, mais plutôt se développer à l'intérieur des parois du canal biliaire commun.
Chez les patients atteints de jaunisse, il peut être nécessaire de placer un stent (un petit tube en plastique ou en métal) pour soulager le canal cholédoque bloqué et résoudre la jaunisse. Cela se fait généralement par une procédure endoscopique appelée CPRE (cholangiopancréatographie rétrograde endoscopique). La procédure comprend deux phases distinctes :
Phase diagnostique : un très petit cathéter est guidé par endoscopie à travers l'ampoule de Vater dans le canal biliaire commun. Une petite quantité d'agent de contraste est ensuite injectée, et des radiographies sont prises. Ce colorant délimite le canal biliaire et le canal pancréatique, et montre le site et la morphologie de l'obstruction biliaire qui pourrait être due à un cholangiocarcinome.
Phase opératoire : une petite incision est pratiquée sur l'orifice ampullaire (papillotomie), puis le stent est passé à travers l'endoscope et est placé dans le canal biliaire. Le stent aide à maintenir le canal cholédoque ouvert et résiste à la compression du cancer environnant.
Au cours d'une CPRE, il est possible d'effectuer une biopsie par brossage du canal cholédoque pour vérifier la présence de cellules néoplasiques. Ce type de biopsie est cependant peu précis. La CPRE est une procédure invasive qui est associée à un profil de complication spécifique, notamment une pancréatite aiguë sévère. De plus, la canulation ampullaire peut être difficile, et la mise en place d'un fil-guide et d'un stent peut échouer si le canal biliaire est petit ou tortueux. Dans de tels cas, la jaunisse peut être résolue en plaçant un drainage biliaire transhépatique percutané (PTBD). Un mince cathéter est placé par voie percutanée dans les voies biliaires intrahépatiques jusqu'à la voie biliaire commune.
L'imagerie confirme le diagnostic et est nécessaire pour mettre en évidence le tumeur. Un système de classification est un moyen standardisé par lequel l'équipe de soins du cancer décrit l'étendue de la propagation d'un cancer et contient différents éléments d'information, notamment
- La taille de la tumeur primaire
- si la tumeur s'est propagée aux organes ou vaisseaux voisins
- si la tumeur s'est propagée aux ganglions lymphatiques voisins
- si la tumeur s'est propagée (métastasée) à des organes éloignés
Le bilan diagnostique du cholangiocarcinome distal est similaire à celui de l'adénocarcinome canalaire pancréatique.
Voici les grandes lignes du test d'imagerie couramment utilisé :
- Tomodensitométrie (CT) à contraste élevé. Le CT scan est un test à rayons X (il utilise des radiations ionisantes) qui produit des images détaillées en coupe transversale du corps. Les logiciels de reconstruction 3D permettent de caractériser la tumeur de manière détaillée. Les scanners CT montrent le canal cholédoque et le pancréas assez clairement et peuvent souvent confirmer l'emplacement de la tumeur. Les scanners CT peuvent également montrer les organes proches, ainsi que les ganglions lymphatiques et les organes éloignés où le cancer pourrait s'être propagé.
- L'imagerie par résonance magnétique (RMN). L'RMN utilise des ondes radio et des aimants puissants au lieu de rayons X. Il s'agit d'une modalité d'imagerie multiplanaire. L'IRM avec cholangio-pancréatographie est très utile pour décrire l'anatomie du système biliaire et la morphologie des rétrécissements des voies biliaires.
- Échographie écoendoscopique (EUS). L'échographie endoscopique est réalisée à l'aide d'une sonde à ultrasons fixée à l'extrémité d'un endoscope. Cela permet une vision directe du duodénum et de la région papillaire ainsi qu'une échographie très détaillée du pancréas et du canal biliaire commun intrapancréatique. Il est probablement préférable à la tomodensitométrie pour repérer un épaississement néoplasique de la paroi du canal cholédoque. Si une tumeur est détectée, une biopsie transgastrique ou transduodénale peut être effectuée au cours de cette procédure.
- Tomographie par émission de positrons (PET). La PET implique l'utilisation d'une très faible dose d'un radiotraceur intraveineux (connu sous le nom de 18-fluorodésoxyglucose ou FDG). Les cellules néoplasiques absorbent une grande quantité de FDG, qui est détectée par le scanner. Ces images fonctionnelles ont cependant une résolution spatiale insuffisante ; c'est pourquoi la TEP-scan est combinée à la tomodensitométrie (PET-CT) pour fournir des images détaillées de la tumeur primaire. La PET-CT peut être particulièrement utile pour repérer un cancer qui s'est propagé au-delà du canal cholédoque et des organes voisins.
Bien que la résolution spatiale des modalités d'imagerie en coupe transversale se soit améliorée, il peut être difficile de distinguer un cholangiocarcinome distal (canal biliaire intrapancréatique) d'un adénocarcinome canalaire de la tête pancréatique. Ce dernier type de cancer peut s'infiltrer de manière diffuse dans le canal cholédoque commun, de sorte qu'il n'est pas possible d'établir d'où la tumeur est apparue.
Le principal marqueur tumoral mesuré dans les néoplasmes pancréatobiliaires est le Ca 19.9, un antigène libéré par les cellules néoplasiques. Les taux sériques de Ca 19,9 sont élevés chez de nombreux patients atteints de cholangiocarcinome distal. Cependant, il existe également des affections non cancéreuses qui provoquent un taux élevé de Ca 19,9, comme les calculs biliaires, la pancréatite, la mucoviscidose, les maladies du foie, les maladies pulmonaires et thyroïdiennes. De plus, le taux de Ca 19,9 peut être élevé chez les personnes souffrant d'une obstruction des voies biliaires, ce qui est le cas de nombreux patients atteints de cholangiocarcinome. Au contraire, chez les patients qui n'ont pas l'antigène Lewis (une protéine du groupe sanguin sur les globules rouges), ce qui représente environ 10% de la population caucasienne, le Ca19.9 n'est pas exprimé, même chez ceux qui ont de grosses tumeurs. C'est pourquoi le Ca 19.9 n'est pas particulièrement utile comme test de diagnostic pour les néoplasmes pancréatobiliaires. Une fois le diagnostic de cholangiocarcinome confirmé, et si le taux de Ca 19,9 de l'individu était élevé avant le traitement, le test Ca 19,9 peut être utilisé comme facteur de pronostic.
Une fois la caractérisation radiologique obtenue (taille de la tumeur, relation avec les vaisseaux péripancréatiques, état des ganglions lymphatiques, présence de métastases), ces informations sont combinées pour attribuer un stade. La classification actuelle est basée sur le système TNM (tumeur/état ganglionnaire/métastase), selon l'American Joint Commitee on Cancer (AJCC, www.cancerstaging.org). Le stade de la tumeur est exprimé en chiffres romains I à IV.
Voici les groupes de stades de l'AJCC pour le cholangiocarcinome extra-hépatique (septième édition, 2010):
- Stade 0 : La tumeur est confinée dans la couche interne du canal biliaire.
- Stade IA : La tumeur est confinée à l'intérieur de la paroi du canal biliaire.
- Stade IB : La tumeur se développe à l'extérieur du canal biliaire. Elle ne s'est pas étendue aux ganglions lymphatiques voisins ou à des sites éloignés.
- Stade IIA : La tumeur s'est propagée aux organes voisins (pancréas, vésicule biliaire, foie). Elle ne s'est pas propagée aux ganglions lymphatiques voisins ou à des sites distants.
- Stade IIB : la tumeur est soit confinée dans le canal biliaire, soit se développe en dehors de celui-ci, mais elle s'est propagée aux ganglions lymphatiques voisins. La tumeur ne s'est pas propagée à des sites distants.
- Stade III : La tumeur se développe à l'extérieur du canal biliaire dans les principaux vaisseaux sanguins. Elle peut ou non s'être propagée aux ganglions lymphatiques voisins. Elle ne s'est pas étendue à des sites distants.
- Stade IV : Le cancer s'est propagé à des sites éloignés.
La classification radiologique divise le cholangiocarcinome distal en groupes selon qu'il est probable ou non qu'il puisse être enlevé chirurgicalement:
- Tumeur résécable
- Tumeur localement avancé
- Tumeur métastatique
La tumeur opérable ou potentiellement opérable
L'approche thérapeutique du cholangiocarcinome distal résécable (stade 0, IA, IB, IIA, IIB) est similaire à celle de l'adénocarcinome canalaire de la tête du pancréas. L'opération de choix est une pancréaticoduodénectomie potentiellement curative avec lymphadénectomie régionale. La résection pancréatique avec résection de la veine porte synchrone, la résection du canal cholédoque commun au niveau de l'hépatite portale et la résection du foie doivent être effectuées dans certains cas.
Bien que les progrès récents de l'imagerie en coupe transversale permettent une évaluation détaillée du canal cholédoque, du pancréas et du foie, la précision de la stadification radiologique n'est pas de 100 %. Lorsqu'il y a de bonnes chances que la tumeur puisse être complètement enlevée, on procède à une intervention chirurgicale, et l'exploration chirurgicale joue toujours le rôle clé pour l'évaluation finale de la résectibilité. Parfois, on découvre une maladie inattendue localement avancée ou une maladie métastatique. Dans ce cas, le chirurgien peut poursuivre l'opération en tant que procédure palliative pour soulager ou prévenir les symptômes.
Toutefois, l'anatomie de la tumeur n'est pas le seul élément à prendre en compte. D'autres facteurs liés au patient sont tout aussi importants pour le processus de décision. Parmi les plus importants, citons les comorbidités et l'état fonctionnel. Les comorbidités font référence à d'autres maladies dont le patient peut souffrir, telles que les maladies cardiaques ou le diabète. L'état fonctionnel fait référence à l'état nutritionnel, à la capacité de subir une opération majeure et de fonctionner de manière autonome après l'opération. L'évaluation de la résectibilité nécessite donc une évaluation complexe de l'anatomie de la tumeur, de l'âge, des comorbidités, de l'état fonctionnel et des résultats d'une analyse sanguine (Ca 19.9) pour déterminer le profil risque-bénéfice de l'opération.
L'objectif de la pancréaticoduodénectomie est d'éliminer complètement la tumeur, sans laisser de résidu cancéreux. Ce concept est connu sous le nom de chirurgie R0 (sans maladie résiduelle). En outre, une lymphadénectomie régionale est toujours pratiquée. Le rôle de la dissection étendue des ganglions lymphatiques est controversé et ne semble pas être bénéfique. Après la résection, une rection congelée peropératoire du canal cholédoque commun et des marges de résection pancréatique est effectuée pour éliminer la maladie microscopique résiduelle (R1) dans le reste du pancréas. L'extension de la résection à la voie supérieure du canal biliaire, jusqu'à la porta-hépatite, ou au corps pancréatique jusqu'à la pancréatectomie totale avec splénectomie peut être nécessaire lorsque les marges de résection sont positives pour les cellules tumorales.
L'échantillon de résection est examiné par le pathologiste. L'examen histologique confirme le diagnostic et attribue le stade pathologique de la tumeur (selon le système TNM, AJCC, www.cancerstaging.org). Bien qu'elles ne fassent pas officiellement partie du système TNM, d'autres caractéristiques histologiques sont d'une importance primordiale. Le grade du cancer (l'aspect anormal des cellules sous le microscope) est indiqué sur une échelle allant de G1 à G4, les cancers G1 ressemblant le plus aux cellules normales et offrant les meilleures perspectives. Comme on l'a vu, un autre facteur important est la clairance de la tumeur, que la tumeur soit entièrement enlevée ou non. Ce facteur est indiqué sur une échelle allant de R0 (où toute la tumeur visible et microscopique a été enlevée) à R2 (où une partie de la tumeur visible n'a pas pu être enlevée). En outre, les ganglions lymphatiques sont analysés pour rechercher une implication du cancer (statut des ganglions lymphatiques).
Les patients reçoivent une chimiothérapie ou une chimio-radiothérapie après l'ablation du cancer afin d'essayer d'éliminer les cellules cancéreuses qui ont été laissées sur place. Ce type de traitement, appelé traitement adjuvant, réduit le risque de récidive du cancer.
Le Tumeur Localement avancé
Le cholangiocarcinome distal est défini comme localement avancé (stade III) lorsqu'il s'est étendu à la veine porte et à l'artère hépatique, ou lorsqu'il s'est étendu à l'artère mésentérique supérieure.
Lorsque l'imagerie en coupe transversale révèle un cholangiocarcinome distal localement avancé, il est nécessaire d'obtenir un diagnostic pathologique (généralement cytologique). Ce diagnostic doit être obtenu par une biopsie par aspiration à l'aiguille fine. Pour ce test, une fine aiguille est insérée à travers la peau et dans la tumeur. L'échographie est utilisée pour examiner la position de l'aiguille et s'assurer qu'elle est bien dans la tumeur. Les biopsies peuvent également être effectuées à l'aide d'ultrasons endoscopiques, en plaçant l'aiguille directement à travers la paroi de l'estomac ou à travers le duodénum dans la tumeur. Dans les deux cas, de petits échantillons de tissu peuvent être prélevés à l'aide de l'aiguille. Si une CPRE est effectuée, une biopsie par brossage peut être obtenue. Les échantillons de tissus sont ensuite examinés au microscope. Les cellules anormales sont trouvées et examinées par le pathologiste, qui décide d'un diagnostic.
Si le tumeur a causé des symptômes, un traitement palliatif peut être nécessaire pour les soulager :
Un stent en plastique ou en métal peut être placé par voie endoscopique pour soulager la jaunisse causée par l'obstruction du canal cholédoque. Une autre solution consiste à mettre en place un drainage biliaire percutané. Pour plus d'informations, cliquez ici.
Si nécessaire, la chirurgie peut rediriger le flux de bile du canal cholédoque directement dans l'intestin grêle, en contournant la zone obstruée (opération de pontage). La connexion de l'estomac au duodénum peut également être redirigée à ce moment-là pour soulager ou prévenir l'obstruction du duodénum. En outre, des biopsies peropératoires peuvent être effectuées.
Le traitement de première ligne du cholangiocarcinome distal localement avancé est la thérapie médicale. La chimiothérapie implique l'utilisation de médicaments pour tuer les cellules cancéreuses, et peut être administrée par voie intraveineuse ou orale. Ces médicaments sont généralement administrés par cycles, avec une alternance de périodes de traitement et de récupération, et peuvent être administrés seuls ou en conjonction avec la radiothérapie (chimio-radiothérapie). L'équipe d'oncologie choisit le meilleur plan de traitement pour chaque patient. Une fois le programme de traitement terminé, on procède à une nouvelle répartition des soins. Cela implique un examen clinique détaillé, la mesure du taux de Ca 19,9 dans le sérum et l'imagerie en coupe (CT-scan ou PET-CT). Les résultats sont discutés au sein de notre équipe multidisciplinaire. Les résultats obtenus après un traitement médical de première ligne pour un cholangiocarcinome distal localement avancé sont les suivants
La régression de la maladie (down-staging). Cela signifie que la maladie est devenue résécable, au moins sur la base de l'imagerie transversale. Le chirurgien peut décider d'une exploration chirurgicale, s'il est probable que le néoplasme puisse être complètement enlevé chirurgicalement.
Maladie stable. Cela signifie qu'aucune nouvelle tumeur ne s'est développée, et que le néoplasme ne s'est pas propagé à de nouvelles régions du corps (en d'autres termes, le néoplasme ne s'améliore pas ou ne s'aggrave pas). Dans une telle situation, la prise en charge est adaptée à chaque patient, après une discussion multidisciplinaire : une chirurgie lourde avec résection vasculaire peut être prise ne considération ou les cas échant une chimiothérapie de deuxième ligne ou une radio-chimiothérapie
Évolution de la maladie. Cela signifie que la tumeur a progressé localement (a pris de l'ampleur) ou s'est étendue à d'autres organes du corps (s'est métastasée). Le site métastatique le plus fréquent est le foie. Dans une telle situation, une chimiothérapie de deuxième ligne peut être indiquée, ainsi que l'inclusion dans des essais cliniques expérimentaux.
Le Tumeur métastatique
Le cancer métastatique est un cancer qui s'est propagé, par la circulation sanguine ou le système lymphatique, de l'endroit où il a débuté à un autre endroit du corps. La capacité d'une cellule cancéreuse à se métastaser dépend de ses propriétés individuelles, des propriétés des cellules non cancéreuses, y compris les cellules du système immunitaire, présentes à l'endroit initial, et des propriétés des cellules qu'elle rencontre dans le système lymphatique ou la circulation sanguine et à la destination finale dans une autre partie du corps. Le cholangiocarcinome distal se métastase le plus souvent au niveau du foie et des ganglions lymphatiques distants (stade IV).
Le traitement médical standard du cholangiocarcinome distal métastatique comprend généralement une chimiothérapie. Il semble y avoir des combinaisons intéressantes et potentiellement prometteuses de plusieurs agents médicamenteux de traitement médical conventionnel qui sont en pratique et à l'étude pour le traitement des carcinomes pancréatobiliaires métastatiques. En outre, un large éventail d’approches de traitement médical monomode est actuellement en cours d’essais cliniques. Il s’agit notamment de certaines des thérapies expérimentales les plus récentes qui visent davantage les cibles moléculaires et l’interruption des voies de signalisation génétique.
Les régimes de traitement médical individualisés, adaptés par des oncologues experts, impliquant des agents uniques ou une thérapie combinée, peuvent prolonger la survie et la qualité de vie. Enfin, les essais cliniques restent une option.
Le Suivi
Après une résection radicale, les patients sont inscrits dans un protocole de surveillance strict, comprenant un examen clinique détaillé, la mesure du Ca 19,9 sérique et l'imagerie transversale. De même, les patients localement avancés et métastatiques sont suivis périodiquement pour vérifier une éventuelle réponse au traitement et pour contrôler les symptômes.
3. La Tumeur Duodénale
L'adénocarcinome primaire du duodénum est une maladie rare qui représente entre 0,3 % et 0,5 % de toutes les malignités gastro-intestinales et 10 % des néoplasmes périampullaires. Il semble être plus fréquent dans la zone périampullaire, mais il a également été démontré qu'il survient dans les troisième et quatrième portions duodénales. Le néoplasme commence dans la muqueuse duodénale ou survient dans le contexte d'autres néoplasmes duodénaux bénins préexistants tels que les polypes, la polypose adénomateuse familiale, l'hyperplasie lymphatique, l'hétérotopie gastrique et pancréatique, le syndrome de Peutz-Jeghers. Les autres affections associées à l'adénocarcinome primaire du duodénum comprennent la maladie de Crohn et la maladie de von Recklinghausen, ainsi que la maladie cœliaque.
Voici les principaux symptômes de l'adénocarcinome primaire du duodénum :
- Perte de poids
- Faiblesse
- Douleurs abdominales
- Dyspepsie
- Hémorragie et anémie
- Obstruction duodénale
- Jaunisse (dans les lésions périampullaires)
L'endoscopie supérieure et l'échographie endoscopique sont largement utilisées pour étudier les néoplasmes duodénaux, qui sont la plupart du temps exophytiques. Il est possible d'effectuer des biopsies pour confirmer le diagnostic. La tomodensitométrie est utilisée pour la classification des tumeurs. La classification actuelle est basée sur le système TNM (tumeur/état ganglionnaire/métastase), selon le Comité conjoint américain sur le cancer (AJCC, www.cancerstaging.org). Le stade de la tumeur est exprimé en chiffres romains I à IV.
Voici les groupes de stades de l'AJCC pour les néoplasmes de l'intestin grêle (septième édition, 2010) :
- Stade 0 : Le néoplasme est confiné à la muqueuse duodénale (carcinome in situ).
- Stade I : Le néoplasme est confiné dans la musculeuse de l'intestin grêle. Il ne s'est pas propagé aux ganglions lymphatiques voisins ou à des sites éloignés.
- Stade IIA : Le néoplasme est confiné dans la séreuse (péritoine). Il ne s'est pas propagé aux ganglions lymphatiques voisins ou à des sites distants.
- Stade IIB : le néoplasme s'est développé en dehors de la séreuse et s'est propagé aux organes adjacents. Il ne s'est pas propagé aux ganglions lymphatiques voisins ou à des sites distants.
- Stade IIIA : Le néoplasme s'est développé en dehors de la séreuse et s'est propagé aux organes adjacents : Le néoplasme s'est propagé aux ganglions lymphatiques voisins (<=3) mais pas à des sites distants.
- Stade IIIB : le néoplasme s'est propagé aux ganglions lymphatiques voisins (>3) mais pas à des sites distants.
- Stade IV : Le néoplasme s'est propagé à des sites distants.
La classification radiologique divise l'adénocarcinome duodénal primaire en groupes selon qu'il est probable ou non qu'il puisse être enlevé chirurgicalement.
L'approche thérapeutique de l'adénocarcinome duodénal primaire opérable(stade 0-IIB) est similaire à celle de l'adénocarcinome canalaire de la tête du pancréas. L'opération de choix est une pancréatico-duodénectomie potentiellement curative avec lymphadénectomie régionale. Une résection avec résection de la veine porte synchrone peut être effectuée si nécessaire.
L'adénocarcinome duodénal primaire localement avancé est une tumeur de stade IIB-IIIA/B qui s'avère techniquement non opérable (par exemple en raison d'une infiltration diffuse de l'artère mésentérique supérieure). Si le néoplasme a provoqué des symptômes, un traitement palliatif peut être nécessaire pour les soulager :
En cas de jaunisse, un stent en plastique ou en métal peut être placé par voie endoscopique pour soulager le canal cholédoque obstrué. Une autre solution consiste à mettre en place un drainage biliaire percutané.
Si nécessaire, la chirurgie peut rediriger la connexion de l'estomac vers le duodénum pour soulager ou prévenir l'obstruction du duodénum. Le flux de bile provenant du canal cholédoque peut alors être redirigé directement vers l'intestin grêle, en contournant la zone obstruée (opération de pontage). En outre, des biopsies peropératoires peuvent être effectuées.
Le traitement de première ligne de l'adénocarcinome duodénal primaire non opérable localement avancé est la thérapie médicale. La chimiothérapie implique l'utilisation de médicaments pour tuer les cellules cancéreuses, et peut être administrée par voie intraveineuse ou orale. Ces médicaments sont généralement administrés par cycles, avec une alternance de périodes de traitement et de récupération, et peuvent être administrés seuls ou en conjonction avec la radiothérapie (chimio-radiothérapie). L'équipe d'oncologie choisit le meilleur plan de traitement pour chaque patient. Une fois le programme de traitement terminé, on procède à une nouvelle répartition des soins. Cela implique un examen clinique détaillé, la mesure des marqueurs tumoraux sériques et l'imagerie transversale (CT-scan ou PET-CT). Les résultats sont discutés au sein de notre équipe multidisciplinaire. Si la maladie a été déclassée, une exploration chirurgicale et - si possible - une résection radicale peuvent être tentées.
Le cancer métastatique est un cancer qui s'est propagé, par la circulation sanguine ou le système lymphatique, de l'endroit où il a débuté à un autre endroit du corps. Le cholangiocarcinome distal se métastase le plus souvent au niveau du foie et des ganglions lymphatiques distants (stade IV).
Le traitement médical standard de l'adénocarcinome duodénal primaire métastatique comprend généralement une chimiothérapie. Il semble y avoir des combinaisons intéressantes et potentiellement prometteuses de plusieurs agents médicamenteux de traitement médical conventionnel qui sont en pratique et à l'étude pour le traitement des néoplasmes gastro-intestinaux métastatiques. En outre, il existe un large éventail d'approches de traitement médical monomode actuellement en cours d'essais cliniques. Il s'agit notamment de certaines des thérapies expérimentales les plus récentes qui visent davantage les cibles moléculaires et l'interruption des voies de signalisation génétique. Des schémas de traitement médical individualisés, adaptés par des oncologues experts, impliquant des agents uniques ou une thérapie combinée, peuvent prolonger la survie et la qualité de vie. Enfin, les essais cliniques restent une option.
4. Le GIST duodénale
La tumeur stromale gastro-intestinale (GIST) est un cancer rare qui affecte le tube digestif ou les structures voisines de l'abdomen. Les GIST représentent 1 à 3 % des néoplasmes gastro-intestinaux. Le site le plus fréquent des GIST est l'estomac (environ 55 %), suivi du duodénum et de l'intestin grêle (environ 30 %), de l'œsophage (environ 5 %), du rectum (environ 5 %), du côlon (environ 2 %) et de rares autres sites. La plupart des GIST présentent des mutations de la protéine CD117 (c-kit).
Les GIST duodénales présentent des douleurs abdominales, des saignements et une obstruction duodénale. Ils sont principalement situés dans la deuxième partie du duodénum et n'ont pas tendance à envahir les organes proches, les ganglions lymphatiques ou les organes éloignés. La chirurgie est le pilier de la thérapie des GIST. Si une GIST est petite et est fixée à une section du duodénum suffisamment éloignée de l'ampoule de Vater, il peut être possible d'effectuer une résection segmentaire. Pour les GIST duodénaux qui impliquent les voies biliaires ou qui sont très proches de l'ampoule de Vater, et pour les GIST qui adhèrent aux organes adjacents, une pancréaticoduodénectomie est nécessaire.
Le risque de récidive ou de métastase d'une GIST primaire réséquée chirurgicalement semble être indépendant du type d'opération pratiquée, et peut être prédit sur la base de la taille de la tumeur et du nombre de mitotiques (tableau des risques du NIH). Pour plus d'informations, cliquez ici.
Le mésylate d'imatinib est le premier traitement médicamenteux efficace contre les GIST, et a été le premier traitement moléculaire ciblé commercialisé pour le cancer. Cela signifie que le médicament n'affecte que les cellules qui expriment des cibles très spécifiques, contrairement aux chimiothérapies anticancéreuses classiques qui affectent toutes les cellules à croissance rapide du corps. Les cibles de l'imatinib chez les patients atteints de GIST sont deux récepteurs de facteur de croissance qui bloquent la protéine c-kit et d'autres protéines kinases qui régulent la prolifération cellulaire. L'imatinib a été initialement approuvé pour les GIST métastatiques. Il a ensuite été utilisé en traitement adjuvant (après une résection radicale), et il est actuellement en cours d'évaluation en thérapie néoadjuvante (pour réduire une tumeur résécable et permettre une résection segmentaire, ou pour réduire les néoplasmes localement avancés). D'autres thérapies moléculaires ciblées ont été récemment approuvées pour le traitement des GIST (sunitinib).
5. Les Tumeur de la région Ampullaire
Géneralités
Les tumeurs ampullaires apparaissent dans l'ampoule de Vater, une structure formée par la coalescence du canal pancréatique et du canal biliaire commun. L'ampoule est spécifiquement située au niveau de la papille duodénale principale, et contrôle l'introduction de la bile et des sécrétions pancréatiques dans le duodénum, tout en empêchant l'entrée du contenu du duodénum dans les conduits.
Les tumeurs périampullaires sont définies comme celles qui surviennent à moins de 2 cm de l'ampoule de Vater. La région périampullaire est histologiquement complexe et comprend différents types épithéliaux, tels que la muqueuse duodénale, le revêtement épithélial du canal biliaire commun et le revêtement épithélial du canal pancréatique principal. Par conséquent, différents types d'adénomes et d'adénocarcinomes peuvent apparaître dans cette zone, notamment l'adénocarcinome du canal pancréatique, le cholangiocarcinome distal, l'adénocarcinome duodénal, ainsi que l'adénocarcinome ampullaire susmentionné.
Les adénocarcinomes ampullaires peuvent être classés en deux sous-groupes histologiques :
- Adénocarcinome ampullaire de type pancréatobiliaire
- Adénocarcinome ampullaire de type intestinal
Jusqu'à 75% des tumeurs survenant dans la région périampullaire sont des adénocarcinomes des canaux pancréatiques. Les cholangiocarcinomes distaux représentent 10 à 40 %, les adénocarcinomes duodénaux sont généralement inférieurs à 10 %, les carcinomes ampullaires représentent 12 à 40 %, selon les différentes séries de cas. Des néoplasmes neuroendocriniens et d'autres néolplasmes mésentésimaux peuvent parfois apparaître dans la région paériamullaire
Tous ces tumeurs présentent des symptômes communs, notamment :
- Douleur abdominale (épigastrique, quadrant supérieur droit)
- Faible appétit
- Faiblesse
- Perte de poids
- La jaunisse
- Pancréatite aiguë
Les adénomes ampullaires et peri-ampullaires
Les néoplasmes bénins de l'ampoule de Vater et de la région périampullaire sont peu fréquents. Il s'agit le plus souvent d'adénomes villosités et tubulo-villosités provenant de l'épithélium ampullaire ou de la muqueuse duodénale. Les adénomes peuvent subir une transformation maligne en carcinomes, similaire à la séquence adénome-carcinome qui se produit ailleurs dans le tractus gastro-intestinal.
Les symptômes sont causés par l'obstruction de l'écoulement ampullaire, qui entraîne un ictère et peut prédisposer à une pancréatite aiguë. L'obstruction du duodénum est moins fréquente.
Malgré leur rareté, les adénomes ampullaires et périampullaires sont de plus en plus reconnus grâce à la disponibilité de l'endoscopie supérieure et de l'échographie endoscopique, ainsi qu'à l'application généralisée des programmes de dépistage et de surveillance pour les patients à haut risque. Les adénomes ampullaires apparaissent sporadiquement et dans le cadre de syndromes de polypose familiale, comme la polypose adénomateuse familiale (PAF). Presque tous les patients atteints de PAF développeront des adénomes ampullaires et des polypes adénomateux duodénaux, qui sont fréquemment nombreux et ont également un potentiel malin.
Les indications pour l'excision d'un adénome ampullaire comprennent le traitement des symptômes immédiats ainsi que la prévention de la dégénérescence maligne. Les polypes duodénaux périampullaires peuvent être excisés par voie endoscopique, de la même manière que les polypes coloniaux.
Les polypes ampullaires peuvent être traités par excision endoscopique locale (ampullectomie endoscopique). Cette procédure est moins invasive que la pancréaticoduodénectomie, présente les avantages d'une morbidité plus faible (0 %-25 %), d'une mortalité essentiellement nulle et d'une durée d'hospitalisation éventuellement réduite, mais des taux de récidive nettement plus élevés (généralement 5 %-30 %) et la nécessité d'une surveillance endoscopique postopératoire. L'ampullectomie peut être pratiquée dans les polypes bénins et dans les lésions présentant une dysplasie/carcinome de haut grade in situ. Comme il a été démontré que les petits carcinomes ampullaires qui se développent en dehors de la muqueuse ampullaire peuvent envahir les ganglions lymphatiques régionaux, l'excision locale peut ne pas être adéquate du point de vue oncologique. Dans de tels cas, la pancréaticoduodénectomie devrait être le traitement de choix.
L’adénocarcinome de l’Ampulle de Vater
L'adénocarcinome de l'ampoule de Vater (ou adénocarcinome ampullaire) se développe dans la région ampullaire, y compris la surface duodénale de l'ampoule, l'épithélium ampullaire de transition et les canaux ampullaires (les extrémités très distales du canal cholédoque et du canal pancréatique), qui sont bordés par l'épithélium pancréatobiliaire.
Le terme "adénocarcinome ampullaire" n'a toujours pas de définition précise, de sorte qu'il n'existe pas de sous-classifications spécifiques pour les tumeurs provenant des différents compartiments de l'ampoule de Vater. Une des subdivisions proposées, basée sur les caractéristiques histologiques et moléculaires de la tumeur, comprend le sous-type intestinal (qui naît de la surface duodénale et engloutit l'orifice papillaire) et le sous-type pancréatobiliaire (qui naît à l'intérieur des extrémités très distales du CBD et/ou du canal pancréatique). Des preuves récentes suggèrent que ces deux sous-types sont associés à un pronostic sensiblement différent, plus favorable dans le sous-type intestinal.
Les adénocarcinomes ampullaires sont généralement diagnostiqués à un stade précoce, car l'engouffrement de l'orifice papillaire provoque une ostruction biliaire et un ictère, ou peut entraîner une pancréatite aiguë. Des masses papillaires peuvent être observées à l'endoscopie supérieure et à l'échographie endoscopique, et une biopsie peut être effectuée.
Chez les patients atteints de jaunisse, il peut être nécessaire de placer un stent (un petit tube en plastique ou en métal) pour soulager le canal cholédoque bloqué et résoudre la jaunisse. Cela se fait généralement par une procédure endoscopique appelée CPRE (cholangiopancréatographie rétrograde endoscopique). La procédure comprend deux phases distinctes :
Phase diagnostique : un très petit cathéter est guidé par endoscopie à travers l'ampoule de Vater dans le canal biliaire commun. Une petite quantité d'agent de contraste est ensuite injectée, et des radiographies sont prises. Ce colorant délimite le canal biliaire et le canal pancréatique, et montre le site et la morphologie de l'obstruction biliaire qui pourrait être due à un cholangiocarcinome.
Phase opératoire : une petite incision est pratiquée sur l'orifice ampullaire (papillotomie), puis le stent est passé à travers l'endoscope et est placé dans le canal biliaire. Le stent aide à maintenir le canal cholédoque ouvert et résiste à la compression du cancer environnant.
Au cours d'une CPRE, il est possible d'effectuer une biopsie par brossage du canal cholédoque pour vérifier la présence de cellules néoplasiques. Ce type de biopsie est cependant peu précis. La CPRE est une procédure invasive qui est associée à un profil de complication spécifique, notamment une pancréatite aiguë sévère. De plus, en raison de la présence de la tumeur, la canulation ampullaire peut être difficile, et la mise en place du fil-guide et du stent peut échouer. Dans de tels cas, la jaunisse peut être résolue en plaçant un drainage biliaire transhépatique percutané (PTBD). Un mince cathéter est placé par voie percutanée dans les voies biliaires intrahépatiques jusqu'à la voie biliaire commune.
L'échographie écoendoscopique (EUS) est très utile pour évaluer les néoplasmes ampullaires. L'échographie endoscopique est réalisée à l'aide d'une sonde à ultrasons qui est fixée à l'extrémité d'un endoscope. Cela permet une vision directe de la région papillaire ainsi qu'une échographie très détaillée de l'ampoule et des tissus environnants. Elle est probablement meilleure que la tomodensitométrie pour l'évaluation de l'ampoule de Vater. En outre, des biopsies peuvent être effectuées au cours de cette procédure.
L'imagerie transversale confirme le diagnostic et est nécessaire pour mettre en évidence le néoplasme. Un système de stadification est un moyen standardisé par lequel l'équipe de soins du cancer décrit l'étendue de la propagation d'un cancer et contient différents éléments d'information, notamment
- la tomographie assistée par ordinateur (CT) à contraste élevé. Le CT scan est un test à rayons X (il utilise des radiations ionisantes) qui produit des images détaillées en coupe transversale du corps. Les logiciels de reconstruction 3D permettent de caractériser la tumeur de manière détaillée. Les scanners CT montrent la région ampullaire, le duodénum et le pancréas assez clairement. Un signe caractéristique du cancer ampullaire est la dilatation simultanée du canal biliaire commun et du canal pancréatique, connue sous le nom de "signe du double canal". Les scanners peuvent également montrer les organes proches, ainsi que les ganglions lymphatiques et les organes éloignés où le cancer pourrait s'être propagé
- L'imagerie par résonance magnétique (IRM). L'IRM utilise des ondes radio et des aimants puissants au lieu de rayons X. Il s'agit d'une modalité d'imagerie multiplanaire. L'IRM avec cholangio-pancréatographie est très utile pour tracer l'anatomie du système canalaire biliaire et pancréatique. Le double signe des canaux est bien visible sur la cholangio-pancréatographie.
- Échographie avec renforcement du contraste (CEUS). Il s'agit d'une nouvelle technique d'imagerie qui implique l'utilisation de microbulles de contraste pour montrer en temps réel les informations sur la perfusion des tissus. Les marges et la taille de la lésion sont plus visibles, ce qui améliore la détection des infiltrations vasculaires. En outre, CEUS améliore la stadification hépatique.
Le principal marqueur tumoral mesuré dans les néoplasmes pancréatobiliaires est le Ca 19.9, un antigène libéré par les cellules néoplasiques. Les taux sériques de Ca 19,9 sont élevés chez de nombreux patients atteints d'adénocarcinome ampullaire. Cependant, il existe également des affections non cancéreuses qui provoquent un taux élevé de Ca 19,9, comme les calculs biliaires, la pancréatite, la mucoviscidose, les maladies du foie, les maladies pulmonaires et thyroïdiennes. De plus, le taux de Ca 19,9 peut être élevé chez les personnes souffrant d'une obstruction des voies biliaires, ce qui est le cas de nombreux patients atteints d'adénocarcinome ampullaire. Au contraire, chez les patients qui n'ont pas l'antigène Lewis (une protéine du groupe sanguin sur les globules rouges), ce qui représente environ 10% de la population caucasienne, le Ca19.9 n'est pas exprimé, même chez ceux qui ont de grosses tumeurs. C'est pourquoi le Ca 19.9 n'est pas particulièrement utile comme test de diagnostic pour les néoplasmes pancréatobiliaires. Une fois le diagnostic d'adénocarcinome ampullaire confirmé, et si le taux de Ca 19,9 de l'individu était élevé avant le traitement, le test Ca 19,9 peut être utilisé comme facteur de pronostic.
Une fois la caractérisation radiologique obtenue (taille de la tumeur, relation avec les vaisseaux péripancréatiques, état des ganglions lymphatiques, présence de métastases), ces informations sont combinées pour attribuer un stade. La classification actuelle est basée sur le système TNM (tumeur/état ganglionnaire/métastase), selon l'American Joint Commitee on Cancer (AJCC, septième édition, 2010, www.cancerstaging.org). Le stade de la tumeur est exprimé en chiffres romains I à IV.
La plupart des adénocarcinomes ampullaires se prêtent à une résection curative. Comme on peut le voir, ces néoplasmes sont diagnostiqués à un stade précoce, lorsqu'ils sont suffisamment éloignés des tissus et des vaisseaux environnants. La procédure de choix est la pancréaticoduodénectomie. Les patients reçoivent une chimiothérapie ou une chimio-radiothérapie après que le cancer a été chirurgicalement enlevé pour essayer d'éliminer les cellules cancéreuses qui ont été laissées derrière eux. Le schéma de chimiothérapie est adapté au sous-type histologique. Ce type de traitement est appelé traitement adjuvant et réduit le risque de récidive du cancer. Le traitement des néoplasmes localement avancés est similaire à celui de l'adénocarcinome canalaire pancréatique et du cholangiocarcinome distal.
L'adénocarcinome ampullaire peut se propager (métastaser) au foie. Le traitement médical standard de l'adénocarcinome ampullaire métastatique comprend une chimiothérapie. Des schémas thérapeutiques individualisés, adaptés par des oncologues experts, impliquant des agents uniques ou une thérapie combinée, peuvent prolonger la survie et la qualité de vie. Enfin, les essais cliniques restent une option.
Après une résection radicale, les patients sont inscrits dans un protocole de surveillance stricte, comprenant un examen clinique détaillé, la mesure du Ca 19,9 sérique et une imagerie transversale. De même, les patients localement avancés et métastatiques sont suivis périodiquement pour vérifier une éventuelle réponse au traitement et pour contrôler les symptômes.
6. Les Tumeur Kystiques du pancréas
Généralités
Bien que considérés comme rares, les néoplasmes kystiques du pancréas sont de plus en plus souvent diagnostiqués grâce à l'utilisation généralisée de l'imagerie en coupe transversale. En fait, dans les centres de soins tertiaires ayant une expérience de la chirurgie du pancréas, la proportion de résections pancréatiques réalisées pour des néoplasmes kystiques a doublé au cours des deux dernières décennies ; parallèlement, le nombre de patients inscrits dans les protocoles de surveillance a considérablement augmenté.
Les néoplasmes kystiques du pancréas englobent un large spectre de lésions bénignes, malignes et limites. De nombreux aspects de leur comportement biologique ont été récemment clarifiés, bien que la compréhension de l'histoire naturelle des formes mucineuses - et en particulier des néoplasmes mucineux papillaires intraconduits de branche - soit limitée par la difficulté de distinguer avec précision les lésions bénignes, malignes et potentiellement malignes avant la résection chirurgicale. En outre, les lignes directrices actuelles pour la prise en charge des néoplasmes kystiques pancréatiques reposent sur l'hypothèse que ces lésions peuvent être classées correctement sur la base de leurs caractéristiques d'imagerie en coupe transversale. Toutefois, il existe un certain degré de chevauchement morphologique entre les différentes lésions, de sorte que la possibilité d'une caractérisation préopératoire inexacte doit toujours être prise en compte. Le diagnostic des néoplasmes kystiques pancréatiques nécessite à la fois une bonne connaissance du spectre morphologique de ces lésions et la collaboration entre chirurgiens, radiologues, gastro-entérologues et pathologistes, afin d'augmenter la probabilité d'une prise en charge appropriée. Cependant, certains aspects de la gestion des néoplasmes kystiques pancréatiques restent flous et, en particulier pour les néoplasmes mucineux, le bilan clinique et radiologique n'est pas toujours en mesure de prédire la probabilité de progression vers un cancer invasif chez un patient donné.
Cela a suscité des controverses quant à savoir si les patients doivent se voir proposer une résection ou s'ils doivent être inscrits dans des protocoles de surveillance avec imagerie périodique. C'est une question pertinente car ces néoplasmes sont le plus souvent diagnostiqués chez des patients asymptomatiques qui ont subi une imagerie transversale pour des problèmes sans rapport. Parmi les nombreux autres aspects non résolus, on peut citer le calendrier approprié de la surveillance, le rôle de l'analyse et de la cytologie des fluides kystiques, le rôle des résections atypiques et de la lymphadénectomie, ainsi que le taux de récurrence des TIPMP et leur association avec d'autres néoplasmes non pancréatiques.
Les Tumeurs Kystiques séreux
Les néoplasmes kystiques séreux sont plus fréquents chez les femmes d'âge moyen. Toute partie de la glande pancréatique peut être affectée. Il s'agit presque toujours de lésions bénignes, car seul un très petit nombre de variantes malignes (cystoadénocarcinomes séreux) a été décrit.
La majorité des néoplasmes kystiques séreux sont asymptomatiques et donc découverts de manière fortuite. Lorsqu'ils sont présents, le symptôme le plus fréquent est une gêne abdominale ou une douleur de faible intensité. La perte de poids, la masse palpable, la jaunisse et la pancréatite obstructive sont peu fréquentes.
Le diagnostic est facile lorsque la lésion présente le motif microkystique caractéristique (en forme de nid d'abeille), consistant en de multiples kystes mesurant 2 cm ou moins séparés par des cloisons fibreuses qui peuvent se fondre en une cicatrice centrale. La variante oligocystique (petit nombre de kystes de plus de 2 cm) et la variante macrokystique (kyste unique) ne sont pas correctement caractérisées par l'imagerie en coupe transversale et peuvent représenter une source d'incertitude diagnostique. Dans ces cas, la différenciation avec les néoplasmes producteurs de mucine peut être difficile. Parfois, les kystes peuvent être minuscules, de sorte que le néoplasme semble radiologiquement solide.
L'imagerie par résonance magnétique avec la cholangio-pancréatographie est la modalité d'imagerie la plus précise ; en revanche, l'échographie endoscopique semble être la seule technique capable de fournir des informations supplémentaires lorsque le diagnostic n'est pas clair. À cet égard, l'analyse du liquide kystique pour le CEA aide à différencier les kystes mucineux des kystes séreux.
La résection est généralement effectuée :
- chez les patients symptomatiques
- lorsque la tumeur ne peut être distinguée des néoplasmes kystiques producteurs de mucine
- dans les lésions très importantes ou à croissance rapide
Chez les candidats à la chirurgie, des résections peu invasives et épargnant le parenchyme peuvent être proposées. Pour plus d'informations sur la chirurgie pancréatique mini-invasive, cliquez ici.
Les lésions asymptomatiques bien documentées peuvent être gérées de manière non chirurgicale.
Les Tumeurs Kystiques mucineux
Les tumeurs kystiques mucineux se rencontrent presque exclusivement chez les femmes d'âge moyen et se situent de préférence dans le corps et la queue du pancréas. Ces lésions ont un potentiel malin. L'âge du patient à la présentation semble dépendre du degré de malignité du néoplasme. Ainsi, les patients atteints de NCM malin sont généralement plus âgés, ce qui suggère une dégénérescence du néoplasme liée au temps à partir d'une lésion initialement bénigne. À l'examen pathologique, une même tumeur peut présenter simultanément tous les différents degrés de transformation maligne.
Un diagnostic précoce des néoplasmes kystiques mucineux est essentiel car le pronostic pour les patients atteints de la forme maligne est le même que pour ceux atteints d'adénocarcinome canalaire, alors que pour les patients présentant des lésions bénignes et un carcinome "in situ", la chirurgie pourrait être curative.
Comme d'autres néoplasmes kystiques, les néoplasmes kystiques mucineux sont le plus souvent diagnostiqués de manière fortuite. Lorsqu'ils sont présents, les symptômes sont non spécifiques et comprennent une gêne ou une douleur abdominale. Il peut également y avoir des symptômes non spécifiques suggérant une malignité, tels que la perte de poids, l'anorexie et la jaunisse obstructive.
Deux types de néoplasmes kystiques mucineux sont observés lors des procédures d'imagerie diagnostique : macrokystique multiloculaire et macrokystique uniloculaire. De fines cloisons délimitent les espaces kystiques, et les calcifications sont fréquentes. Une taille importante (>4 cm), une paroi épaissie, la présence de projections papillaires provenant de la paroi ou de septa, la présence de calcifications périphériques et l'invasion des structures vasculaires environnantes sont considérés comme des signes de malignité.
Dans la mesure du possible, tous les néoplasmes kystiques mucineux doivent être réséqués. Dans les lésions ne présentant pas de signes de malignité, des résections peu invasives et épargnant le parenchyme peuvent être proposées. Pour plus d'informations sur la chirurgie pancréatique mini-invasive, cliquez ici. Dans les lésions inquiétantes ou ouvertement malignes, une résection pancréatique formelle doit être effectuée. Les patients présentant de petites lésions qui ne sont pas chirurgicalement aptes peuvent être surveillés, mais leur issue à long terme est inconnue.
Les Tumeurs intracanalaires papillaires et mucineuses du pancréas (TIPMP)
Les tumeurs intracanalaires papillaires et mucineuses du pancréas (TIPMP) ont été signalés pour la première fois par Ohashi en 1982. Ils sont généralement diagnostiqués dans la sitxième décennie de la vie, tant chez les hommes que chez les femmes. Les TIPMP se caractérisent par la prolifération de cellules mucineuses formant une projection papillaire dans le canal pancréatique principal et/ou ses canaux secondaires. Il en résulte une dilatation kystique des conduits concernés, qui contiennent souvent des bouchons de mucine.
Quatre sous-types épithéliaux histologiques de la TIPMP ont été décrits :
- Type gastrique
- Type d'intestin
- Type pancréatobiliaire
- Type oncocytaire
Le TIPMP englobe un large spectre de comportements biologiques, allant de bénins à ouvertement malins. La classification de l'OMS comprend :
- Lésions bénignes (displasie de bas grade)
- Lésions limites (displasie de grade intermédiaire/supérieur)
- Lésions malignes (carcinome invasif)
- Les carcinomes invasifs sont sous-classés, en fonction de leur type histologique, en
- carcinome tubulaire TIPMP, provenant de l'épithélium gastrique et pancréatobiliaire
- Carcinome colloïdal TIPMP, provenant principalement de l'épithélium intestinal
- TIPMP-carcinome oncocytaire, provenant de l'épithélium oncocytaire
Tout comme les néoplasmes kystiques mucineux, une même tumeur peut présenter simultanément tous les degrés de transformation maligne.
D'un point de vue morphologique, les TIPMP se distinguent en trois entités différentes, selon l'implication canalaire (figure 1). Il a été démontré que cette distinction a une pertinence pronostique :
- TIPMP du conduit principal, impliquant exclusivement le conduit pancréatique principal. Ces néoplasmes sont agressifs, et abritent un carcinome in situ ou invasif dans jusqu'à 70% des spécimens réséqués. La dilatation du canal pancréatique principal peut être segmentaire ou peut concerner le canal entier (IPMN pancréatique)
- TIPMP des conduits secondaires, impliquant exclusivement les conduits secondaires. Ces néoplasmes ont un potentiel malin faible/moyen (jusqu'à 20-25%), et abritent un carcinome invasif chez jusqu'à 10% des spécimens réséqués. Les TIPMP des conduits secondaires sont souvent multifocaux (implication multiple de produits de marque).
- Les TIPMP mixtes, qui impliquent simultanément le conduit principal et les conduits secondaires. Le comportement biologique des TIPMP de type mixte est similaire à celui des TIPMP de conduit principal. La composante des conduits de dérivation est souvent multifocale.
Les Tumeurs kystiques pseudo-papillaires
Les tumeurs kystiques pseudo-papillaires (également connus sous le nom de néoplasmes kystiques solides, papillaires solides, ou tumeur de Franz) sont des néoplasmes malins de bas grade qui se produisent principalement chez les jeunes femmes (>90%) entre 30 et 40 ans. Parmi les néoplasmes kystiques, les néoplasmes pseudopapillaires du pancréas sont les moins fréquents. Selon la classification des tumeurs kystiques de l'OMS, leur origine est incertaine et le comportement biologique est mal défini. Les séries de cas publiées indiquent que les néoplasmes kystiques pseudo-papillaires sont des tumeurs à croissance lente et peu agressive, et même lorsqu'elles sont malignes, elles ont généralement un pronostic favorable. 10 à 15 % de tous les patients auront des métastases, qui sont souvent présentes au moment du premier diagnostic.
Les petits tumeur kystiques pseudo-papillaires sont le plus souvent diagnostiqués de manière fortuite. Lorsque la lésion devient suffisamment importante pour provoquer des symptômes, la douleur abdominale est le symptôme prédominant et, parfois, le seul présent. La douleur peut être associée à une masse abdominale palpable, à une anorexie ou à une perte de poids, mais chacun de ces signes peut se manifester de manière isolée. Ces patients se plaignent généralement d'une sensation de plénitude et d'inconfort abdominal, et ce n'est qu'à l'examen qu'une masse peut être appréciée, en particulier dans le quadrant supérieur gauche.
Sur l'imagerie en coupe, les tumeurs kystiques pseudo-papillaires sont des masses bien vascularisées et encapsulées avec des marges bien définies. Des calcifications et des septa peuvent être observés à l'intérieur de la masse, mais ils ne sont pas pathognomoniques. Au lieu de cela, les résultats distinctifs de ces tumeurs sont l'alternance de zones solides et kystiques, dans lesquelles une composante hémorragique nécrotique peut être présente. Ces observations peuvent être observées dans la même lésion, éventuellement avec des différences dans les proportions des deux composants. L'échographie endoscopique avec biopsie à l'aiguille fine peut aider à confirmer le diagnostic.
Un traitement chirurgical doit être envisagé chez tous les patients chez qui on a diagnostiqué des néoplasmes kystiques pseudo-papillaires, en se basant sur le comportement biologique encore inconnu et la malignité potentielle de ces tumeurs. L'approche laparoscopique s'est avérée sûre et faisable, si l'expertise est disponible. La maladie métastatique n'est pas considérée comme une contre-indication à la chirurgie, car la survie après la résection des métastases hépatiques est favorable. Les récidives sont principalement observées dans les néoplasmes kystiques pseudo-papillaires malins, mais la survie à long terme de ces patients a été signalée s'ils sont traités.
7. Les Tumeurs Endocrines du pancréas
Généralité
Les tumeurs endocrines du pancréas sont un ensemble de types de cellules tumorales collectivement appelées néoplasmes neuroendocriniens du pancréas. Ces tumeurs ont pour origine les cellules des îlots de Langerhans, qui appartiennent au système endocrinien gastro-entéro-pancréatique. Cliquez ici pour plus d'informations sur les cellules des îlots de Langerhans. Il s'agit de néoplasmes peu courants, avec une incidence de 4 à 5 cas pour 100 000 personnes. Cependant, des données récentes indiquent une incidence croissante.
D'un point de vue épidémiologique, les tumeurs neuroendocriniens du pancréas sont divisés en deux groupes :
- Les tumeurs sporadiques. Ils sont plus courants et ne sont pas des maladies néoplasiques héréditaires
- Tumeurs associées à des syndromes génétiques : Ces néoplasmes sont en rapport avec des syndromes génétiques tels que la néoplasie endocrinienne multiple de type 1 (MEN1), le syndrome de Von Hippel-Lindau, la neurofibromatose de type 1 et la sclérose tubéreuse
Les tumeurs neuroendocriniens du pancréas sont cliniquement classés en deux groupes sur la base de la production d'hormones :
- Les tumeurs neuroendocriniens fonctionnels. Les néoplasmes fonctionnels provoquent une augmentation des niveaux d'hormones, et sont associés à des syndromes hormonaux. Les néoplasmes fonctionnels les plus courants sont l'insulinome et le gastrinome
- Tumeurs neuroendocriniens non fonctionnels. Les néoplasmes non fonctionnels ne produisent pas d'hormones associées à des syndromes cliniques. Ils ne provoquent donc pas de symptômes spécifiques, mais plus de 50 % sont malins. Ils sont plus fréquents que les néoplasmes fonctionnels
En outre, les néoplasmes neuroendocriniens du pancréas sont classés sur la base d'un système d'échelle (G1 à G3) proposé par l'Organisation mondiale de la santé (OMS) en 2010. La classification est attribuée par un pathologiste après examen des échantillons de biopsie ou de chirurgie. Le grade de la tumeur est basé sur les caractéristiques morphologiques des cellules néoplasiques, le nombre de mitotiques ou la mesure de l'indice de prolifération Ki-67.
Les tumeurs neuroendocriniens pancréatiques bien différenciés (G1/G2) sont constitués de cellules qui ressemblent beaucoup aux cellules saines du pancréas, et ont un faible taux mitotique ou un faible Ki-67. Les tumeurs bien différenciées sont également moins susceptibles d'être agressives. 90 % des néoplasmes neuroendocriniens du pancréas sont bien différenciés.
Les néoplasmes neuroendocriniens pancréatiques mal différenciés (G3) sont constitués de cellules qui ressemblent moins aux cellules saines du pancréas et présentent un nombre mitotique élevé ou un Ki-67 élevé. Les tumeurs mal différenciées sont agressives.
La Société européenne des tumeurs neuroendocriniennes (ENETS) a proposé un système de classification TNM pour les néoplasmes neuroendocriniens. Le système tumeur/état ganglionnaire/métastase évalue la taille de la tumeur primaire, si la tumeur s'est propagée aux organes ou vaisseaux proches, si la tumeur s'est étendue aux ganglions lymphatiques proches et si la tumeur s'est étendue (métastasée) aux organes éloignés. Il a été démontré que le système de classification ENETS est bien corrélé avec le pronostic du patient, et il est actuellement appliqué dans la pratique clinique.
Bilan diagnostique et classification
Les manifestations cliniques des néoplasmes fonctionnels résultent très souvent des effets métaboliques distinctifs des hormones sécrétées par les cellules néoplasiques, plutôt que du volume de la tumeur ou de la maladie métastatique. Par conséquent, les néoplasmes fonctionnels sont généralement diagnostiqués à un stade précoce, et peuvent même être trop petits pour être détectés par les techniques d'imagerie en coupe. Au contraire, les néoplasmes non fonctionnels ne produisent pas de syndromes cliniques spécifiques (bien qu'ils puissent sécréter des produits aminés et peptidiques inactifs), et ont tendance à se présenter à des stades cliniques ultérieurs avec des symptômes attribuables à un effet de masse ou à des métastases. Cependant, grâce à l'application généralisée de l'imagerie transversale, comme la tomodensitométrie et l'imagerie par résonance magnétique, le nombre de petits néoplasmes neuroendocriniens pancréatiques non fonctionnels découverts accidentellement augmente de façon spectaculaire. Pour en savoir plus sur les symptômes spécifiques des néoplasmes neuroendocriniens, consultez les sections sur les néoplasmes fonctionnels et les néoplasmes non fonctionnels.
Les néoplasmes neuroendocriniens du pancréas apparaissent dans la plupart des cas sous la forme de masses ovales, bien circonscrites et riches en vaisseaux sanguins. Dans une minorité de cas, leurs caractéristiques morphologiques peuvent être atypiques, ce qui rend difficile le diagnostic différentiel avec l'adénocarcinome canalaire. Les néoplasmes neuroendocriniens du pancréas peuvent parfois se présenter sous la forme de néoplasmes kystiques.
Si un néoplasme neuroendocrinien pancréatique est suspecté, une imagerie transversale de haute qualité est nécessaire pour confirmer le diagnostic et mettre en évidence le stade du néoplasme. Un système de stadification est un moyen standardisé par lequel l'équipe de soins du cancer décrit l'étendue de la propagation d'un cancer et contient différents éléments d'information, notamment:
- La taille de la tumeur primaire
- si la tumeur s'est propagée aux organes ou vaisseaux voisins
- si la tumeur s'est propagée aux ganglions lymphatiques voisins
- si la tumeur s'est propagée (métastasée) à des organes éloignés
On décrit ici les tests d'imagerie permettant de diagnostiquer et de mettre en évidence les néoplasmes neuroendocriniens du pancréas.
- Tomodensitométrie (CT) à contraste amélioré. Le CT scan est un test à rayons X (il utilise des radiations ionisantes) qui produit des images détaillées en coupe transversale du corps. Les logiciels de reconstruction 3D permettent de caractériser la tumeur de manière détaillée. Les scans CT montrent le pancréas assez clairement et peuvent souvent confirmer l'emplacement de la tumeur, qui semble hyso-hyperdense et richement mise en valeur. Les scanners peuvent également montrer les organes proches du pancréas, ainsi que les ganglions lymphatiques et le foie, où le cancer pourrait s'être propagé.
- L'imagerie par résonance magnétique (IRM). L'IRM utilise des ondes radio et des aimants puissants au lieu de rayons X. Il s'agit d'une modalité d'imagerie multiplanaire. L'IRM est une modalité d'imagerie complémentaire pour la stadification des néoplasmes neuroendocriniens du pancréas. En particulier, elle peut être très utile pour localiser de petites lésions, grâce à la résolution élevée du contraste. De plus, l'utilisation d'agents de contraste spécifiques à l'hépatite (Gd-BOPTA) permet une meilleure détection des petites métastases hépatiques.
- Échographie à contraste renforcé (CEUS). Il s'agit d'une nouvelle technique d'imagerie qui implique l'utilisation de microbulles de contraste pour montrer en temps réel les informations sur la perfusion des tissus. Dans l'examen CEUS, les néoplasmes neuroendocriniens du pancréas montrent une amélioration précoce et riche pendant les phases dynamiques. Les marges et la taille de la lésion sont plus visibles, ce qui améliore la détection de l'infiltration vasculaire. De plus, le CEUS améliore la stadification hépatique.
- Échographie écoendoscopique (EUS). L'échographie endoscopique est réalisée à l'aide d'une sonde à ultrasons qui est fixée à l'extrémité d'un endoscope. Cela permet une vision directe du duodénum et de la région papillaire ainsi qu'une échographie très détaillée du pancréas, qui se trouve à côté du duodénum. Elle est particulièrement utile, et probablement meilleure que le scanner, pour repérer les petites tumeurs (comme les néoplasmes non fonctionnels diagnostiqués de manière fortuite ou les néoplasmes fonctionnels de la région duodéno-pancréatique). Si une tumeur est détectée, une biopsie trans-gastrique ou trans-duodénale peut être effectuée au cours de cette procédure.
- Scintigraphie des récepteurs de la somatostatine (OCTREOSCAN™). Il s'agit d'un test de médecine nucléaire utilisant un analogue de la somatostatine (octréotide) qui a été lié à une substance radioactive (indium-111). L'octréotide fixe les récepteurs de la somatostatine qui peuvent être exprimés par les cellules néoplasiques de nombreux néoplasmes neuroendocriniens. Après un certain temps après l'injection d'octréotide, une gamma-caméra est utilisée pour montrer où la radioactivité s'est accumulée dans le corps. D'autres scanners peuvent également être effectués les jours suivants. Ce scanner peut aider à diagnostiquer les néoplasmes neuroendocriniens, mais il peut aussi aider à décider d'un traitement. Si un néoplasme neuroendocrinien apparaît sur un site OCTREOSCAN™, cela signifie souvent que la tumeur cessera de croître si elle est traitée avec de l'octréotide. Ce test est moins fréquemment utilisé en raison de l'introduction des radiotraceurs TEP.
- Tomographie par émission de positrons au gallium-68 (TEP-CT 68-Ga). La TEP-scan implique l'utilisation d'une très petite dose d'un radiotraceur intraveineux qui est absorbé par les cellules néoplasiques et détecté par le scanner. Le scanner TEP au Gallium-68 est basé sur une technologie similaire à celle de OCTREOSCAN™. En particulier, le composé marqué au Gallium-68, DOTATATE, se lie très fortement aux récepteurs de la somatostatine exprimés par de nombreuses cellules néoplastiques neuroendocrines. La TEP est combinée à la tomodensitométrie pour fournir des images détaillées. Le scanner TEP/CT au Gallium-68 offre une résolution spatiale plus élevée que les scanners à l'octréotide (3-6 mm contre 10-15 mm), et donne des résultats plus efficaces pour la détection de très petits néoplasmes et métastases.
Une fois la caractérisation radiologique obtenue (taille de la tumeur, relation avec les vaisseaux péripancréatiques, état des ganglions lymphatiques, présence de métastases), ces informations sont combinées pour attribuer un stade. Deux systèmes de classification basés sur la TNM (tumeur/état ganglinnaire/métastase) sont actuellement utilisés. Le premier a été proposé par l'American Joint Commitee on Cancer (AJCC, www.cancerstaging.org), le second par la Société européenne des tumeurs neuroendocrines (ENETS, cliquez ici pour plus d'informations). Le stade de la tumeur est exprimé en chiffres romains I à IV. Voici les groupes de stades ENETS des tumeurs neuroendocriniens du pancréas :
- Stade I : La tumeur est confinée au pancréas et mesure moins de 2 cm. Elle ne s'est pas étendue aux ganglions lymphatiques voisins ou à des sites éloignés.
- Stade IIA : La tumeur est confinée au pancréas et mesure entre 2 et 4 cm. Elle ne s'est pas propagée aux ganglions lymphatiques voisins ou à des sites distants.
- Stade IIB : la tumeur mesure plus de 4 cm et se développe dans le canal cholédoque ou le duodénum. Elle ne s'est pas étendue aux ganglions lymphatiques voisins ou à des sites distants.
- Stade IIIA : La tumeur se développe en dehors du pancréas (estomac, rate, côlon, rein, surrénales) et/ou dans l'artère mésentérique supérieure ou le tronc cœliaque. Elle ne s'est pas étendue aux ganglions lymphatiques voisins ou à des sites distants.
- Stade IIIB : La tumeur s'est propagée aux ganglions lymphatiques voisins mais pas à des sites distants.
- Stade IV : Le cancer s'est étendu à des sites distants.
La classification radiologique divise les néoplasmes neuroendocriniens du pancréas en groupes, selon que le néoplasme est confiné au pancréas, s'est propagé à des organes proches ou s'est étendu à des organes éloignés.
Les tumeurs neuroendocriniens fonctionnels
Insulinome
L'insulinome provient de cellules bêta, situées dans les îlots de Langerhans (cliquez ici pour en savoir plus). Les cellules bêta produisent de l'insuline et une hormone qui régule le métabolisme du sucre. La prolifération des tumeurs entraîne une production d'insuline inappropriée, ce qui se traduit par une hypoglycémie (faible taux de sucre dans le sang). Les caractéristiques cliniques de cette tumeur sont liées à l'hypoglycémie et comprennent la faiblesse, la confusion, la transpiration, un rythme cardiaque rapide, la faim et la prise de poids. Lorsque la glycémie est très basse, le patient peut s'évanouir ou même tomber dans le coma et avoir des convulsions. Ces symptômes sont soulagés par l'alimentation et la consommation de glucose. L'insulinome présente généralement ses premiers symptômes entre 40 et 50 ans, est plus fréquent chez les femmes et a tendance à être de petite taille. Il est en fait le plus souvent diagnostiqué à un stade précoce (<2 cm), car une augmentation légère mais persistante du taux d'insuline suffit à provoquer les symptômes. Les insulinomes sont beaucoup plus susceptibles d'être bénins que malins. Seuls 10 % sont malins, et seulement 10 % sont multiples. Les insulinomes multiples sont généralement observés chez les jeunes patients atteints du syndrome MEN-1. Une hypoglycémie à jeun (<40 mg/dL) associée à un taux d'insuline élevé (en l'absence d'administration exogène d'insuline) est pathognomonique. La mesure de la proinsuline plasmatique peut être utile pour diagnostiquer les carcinomes insulino-sécréteurs. Comme les insulinomes sont généralement de petite taille, dans 20 à 30 % des cas, la tumeur primaire ne peut être détectée par imagerie transversale. Lorsque la tumeur primaire ne peut être évaluée mais que la présence d'un syndrome hormonal lié à l'hypersécrétion d'insuline a été établie, une exploration chirurgicale avec échographie peropératoire est obligatoire. Si l'échographie peropératoire ne permet pas de localiser le néoplasme primaire, une résection chirurgicale "en aveugle" est contre-indiquée. L'excision chirurgicale curative, par laparotomie ou laparoscopie, est le traitement de choix. Dans les lésions sans risque de malignité, des résections épargnant le parenchyme, telles que la pancréatectomie du segment moyen et l'énucléation, peuvent être proposées. Ces opérations peuvent être réalisées par des techniques peu invasives.
Gastrnome
Le gastrinome provient de cellules spécialisées qui fabriquent la gastrine, une hormone qui régule la sécrétion d'acide gastrique. La prolifération de la tumeur entraîne une production inappropriée de gastrine, ce qui entraîne un état connu sous le nom de syndrome de Zollinger-Ellison. Le syndrome de Zollinger-Ellison est caractérisé par une triade de signes et de symptômes, dont l'ulcère gastro-duodénal atypique, l'hyperacidité gastrique et l'hypersécrétion. Les ulcères peuvent se mettre à saigner, entraînant une anémie et une méléna (selles noires et goudronneuses), ou peuvent provoquer une perforation gastrique/duodénale. Les ulcères chez les patients atteints de gastrinomes peuvent être difficiles à traiter, nécessitant de fortes doses de médicaments anti-ulcéreux pour guérir. Les patients doivent prendre ces médicaments pendant longtemps, car les ulcères ont tendance à revenir si le traitement est arrêté. Les autres symptômes sont la diarrhée et la perte de poids. Les gastrinomes sont généralement situés dans le triangle des gastrinomes, qui correspond à peu près à la zone duodéno-pancréatique. La tumeur est souvent > 2cm au moment du diagnostic, elle est multifocale chez 60% des patients, et peut être associée au syndrome MEN1 (20-60%). La plupart des patients sont de sexe masculin (60%) et l'âge moyen au moment du diagnostic est d'environ 60 ans. Plus de la moitié des gastrinomes (60-65%) sont malins, 50% des patients auront des métastases au moment du diagnostic. Le diagnostic dépend de l'élévation du taux de gastrine sérique et du taux d'acide gastrique. Un test provocateur à la sécrétine peut provoquer une élévation substantielle de la gastrine sérique. La tumeur primaire peut ne pas être détectée par l'imagerie transversale, alors que la tomodensitométrie et l'IRM sont très précises pour la détection des métastases hépatiques. La technique d'imagerie la plus précise pour la localisation des gastrinomes est l'échographie endoscopique, qui s'est avérée capable de détecter 50% des lésions duodénales et jusqu'à 75-90% des lésions pancréatiques. La gestion des gastrinomes est chirurgicale. L'approche du traitement dépend souvent des résultats des études de localisation préopératoire (sachant que le gastrinome peut être multifocal), et des résultats de la laparotomie exploratoire. De très petites lésions (< 1cm) situées à la surface du pancréas peuvent être énucléées. Sinon, la pancréaticoduodénectomie avec lymphadénectomie régionale est la procédure de choix. Lorsque la tumeur est située à l'intérieur du duodénum, une duodénotomie avec exploration de la surface interne du duodénum peut être nécessaire. Une échographie peropératoire doit toujours être effectuée pour vérifier la multifocalité.
Le vipome est issu de cellules spécialisées qui fabriquent le peptide intestinal vasoactif (VIP), une hormone qui stimule la sécrétion d'eau et d'électrolytes par l'intestin, la dilatation des vaisseaux sanguins intestinaux et des muscles lisses, et la sécrétion de bicarbonate par le pancréas tout en bloquant la sécrétion d'acide gastrique. Ces phénomènes entraînent une augmentation de la motilité intestinale. La prolifération des tumeurs entraîne une production inappropriée de VIP, ce qui aboutit à un état connu sous le nom de syndrome de Verner-Morrison. Le syndrome de Verner-Morrison se caractérise par une diarrhée aqueuse (jusqu'à 20 selles par jour), une hypokaliémie (faible taux de potassium) et une hypo-/achlorydrie. Une réanimation liquidienne immédiate est souvent nécessaire pour corriger les problèmes électrolytiques et liquidiens (déshydratation, acidose) qui surviennent à la suite du syndrome dont souffrent les patients. Des niveaux élevés de VIP sont diagnostiqués. La tumeur est souvent > 3cm au moment du diagnostic, plus de la moitié des VIPomas (60-65%) sont malins, et situés dans la queue du pancréas. Le traitement de choix est la résection chirurgicale (pancréatectomie gauche). Les analogues de la somatostatine sont également utilisés pour réduire les pertes importantes de liquides et d'électrolytes avant l'intervention chirurgicale. Dans le cas d'une maladie localement avancée ou métastatique, où une résection curative n'est pas possible, il convient d'envisager le débullage et l'élimination de la maladie brute, y compris des métastases, pour atténuer les manifestations caractéristiques de la surproduction de VIP. Les VIPomas sont rarement associés au syndrome MEN1. Occasionnellement, des tumeurs surrénales et des tumeurs des ganglions nerveux, du rétropéritoine et du poumon peuvent provoquer un sydrome de type Verner-Morrison.
Glucagome
Le glucagonome résulte de cellules spécialisées qui fabriquent le glucagon, une hormone pancréatique qui augmente le taux de glucose dans le sang. Son effet est opposé à celui de l'insuline. La prolifération des tumeurs entraîne un excès de glucagon, qui peut faire augmenter le taux de sucre dans le sang, ce qui conduit au diabète. Les patients ont également des problèmes de diarrhée, de perte de poids et de malnutrition (carence en micronutriments). Les problèmes de nutrition peuvent entraîner des symptômes tels que la glossite (irritation de la langue), la chiélite angulaire (irritation aux coins de la bouche) et l'anémie. La caractéristique la plus distinctive du glucagonome est une éruption cutanée appelée érythème migrateur nécrolytique. Il s'agit d'une éruption rouge avec gonflement et cloques et elle se déplace souvent de place en place sur la peau. Ces tumeurs ont tendance à être grosses et facilement visibles au scanner, et sont principalement situées dans la tête et le cou du pancréas. Environ 75 % des glucagonomas sont malins et développent des métastases dans les ganglions lymphatiques et le foie. La résection chirurgicale est le pilier de la thérapie, et une survie prolongée est possible même lorsque la maladie est métastatique. La résection des métastases est également à envisager lorsque cela est possible. La chimiothérapie ou l'octréotide sont utilisés dans les cas de maladie avancée pour soulager les symptômes.
Somatostatinome
Le somatostatinome provient de cellules spécialisées qui fabriquent la somatostatine, une hormone gastro-entéro-pancréatique qui inhibe la production de différentes autres hormones, dont l'insuline, le glucagon, la gastrine et l'hormone de croissance. De plus, la somatostatine inhibe la sécrétion des enzymes pancréatiques, l'excrétion de la bile par la vésicule biliaire, l'absorption des acides aminés et la sécrétion des acides gastriques. La somatostatine inhibe la motilité intestinale et ralentit le temps de transit intestinal. Les somatostatinomes se forment dans la tête pancréatique, dans le duodénum périampullaire, dans l'ampoule de Vater et dans l'intestin grêle. Les somatostatinomes duodénaux sont associés au syndrome de Von Recklinghausen. Les somatostatinomes peuvent également être associés au syndrome MEN1. Ils se présentent souvent sous la forme de diarrhée, de stéatorrhée, de diabète et/ou de calculs biliaires. Une diminution de la sécrétion pancréatique d'enzymes et de bicarbonate explique la diarrhée et la stéatorrhée. L'inhibition de la cholécystokinine par la somatostatine entraîne la formation de calculs biliaires. La somatostatine inhibe également l'insuline, produisant une hyperglycémie. Comme les symptômes d'un somatostatinome sont généralement légers, ces tumeurs ont tendance à être diagnostiquées à un stade avancé. La tomodensitométrie, l'IRM et l'échographie endoscopique peuvent généralement aider à localiser la tumeur et à en déterminer le stade. La plupart de ces tumeurs sont malignes et présentent des métastases au moment du diagnostic (70 %). L'excision complète est la thérapie de choix, si elle est techniquement possible. Cependant, les métastases empêchent souvent une résection curative, et un débulquage palliatif peut être envisagé pour soulager les symptômes. La chimiothérapie est peu efficace.
Autre tumeurs endocrine fonctionnels
Les très rares néoplasmes pancréatiques neuroendocriniens fonctionnels comprennent le corticotropinome, l'ACTHoma, le GRFoma. Ils ne présentent pas de syndrome hormonal spécifique et sont extrêmement difficiles à diagnostiquer. L'excision chirurgicale complète est la seule option curative lorsqu'elle est techniquement possible, et le débulking ou les analogues de la somatostatine sont utilisés pour pallier les syndromes hormonaux.
Syndromes endocrines fonctionnelles sans tumeur visible
Bien que la résolution spatiale des modalités d'imagerie se soit considérablement améliorée, l'insulinome et le gastrinome restent non détectés dans 10 à 20 % des cas, malgré la présence d'un syndrome hormonal bien défini. Dans une telle situation, l'exploration chirurgicale est la seule chance d'identifier la tumeur primaire. Après une laparotomie, il faut procéder à une inspection minutieuse du foie, de l'estomac et du mésentère. La zone duodéno-pancréatique doit être bien exposée et mobilisée à partir du rétropéritoine. L'échographie peropératoire est très utile pour localiser les lésions pancréatiques, avec une précision allant jusqu'à 90-95%. De plus, l'échographie peropératoire est utile pour évaluer la relation entre le néoplasme primaire et le canal pancréatique principal. Cela permet au chirurgien de choisir entre une opération épargnant le parenchyme (énucléation ou pancréatectomie du segment moyen) et une résection formelle (pancréaticoduodénectomie ou pancréatectomie gauche avec dissection des ganglions lymphatiques régionaux). Malheureusement, l'échographie peropératoire est moins précise pour localiser les gastrinomes duodénaux (30%). Lorsque la tumeur est située à l'intérieur de la paroi duodénale, une duodénotomie avec exploration de la surface interne du duodénum est nécessaire. Si le néoplasme primaire reste non détecté, la résection en aveugle est contre-indiquée. Le patient doit plutôt être inscrit dans un protocole de surveillance clinique et radiologique strict, et le syndrome hormonal doit être contrôlé par un traitement médical. Si la tumeur primaire est détectée et qu'une résection est entreprise, l'échantillon doit être analysé par le pathologiste pour confirmer la présence de la tumeur.
Les Tumeurs Endocrines non-fonctionnelles
Les tumeurs neuroendocriniens pancréatiques non fonctionnels ne produisent pas d'hormones actives et ne sont pas associés à des syndromes cliniques spécifiques. Ils peuvent sécréter des produits aminés et peptidiques inactifs, tels que la neurotensine, l'énolase spécifique aux neurones, le polypeptide pancréatique et la chromogranine A. La plupart des lésions sont bien différenciées (G1/G2 selon la classification de l'Organisation mondiale de la santé de 2010), à croissance lente, et se présentent à des stades cliniques ultérieurs avec des symptômes attribuables à un effet de masse, tels que la compression ou l'infiltration d'organes voisins. Une proportion assez élevée de patients (entre 35 et 70 %) aura des métastases au moment du diagnostic, éventuellement au foie. Comme les cellules tumorales peuvent se propager via le système sanguin et lymphatique, il peut y avoir des métastases dans d'autres zones du corps comme les ganglions lymphatiques et les os.
Ces néoplasmes sont généralement diagnostiqués entre 40 et 60 ans, et sont généralement plus importants que leur équivalent fonctionnel. Toutefois, en raison de la disponibilité généralisée de l'imagerie (CT-scan, imagerie par résonance magnétique), les petits tumeurs neuroendocriniens pancréatiques non fonctionnels et asymptomatiques sont identifiés plus fréquemment lors d'examens effectués pour d'autres raisons. Entre 20 et 40 % de ces néoplasmes sont associés à l'HEM1.
Les symptômes les plus fréquents lors de la présentation de néoplasmes neuroendocriniens pancréatiques non fonctionnels sont les suivants
- Douleurs abdominales
- Perte de poids
- Anorexie et nausées
- Hémorragie gastro-intestinale
- La jaunisse
- Masse palpable
Si un néoplasme neuroendocrinien pancréatique non fonctionnel est suspecté sur la base d'une imagerie transversale, le sang peut être testé pour certains marqueurs tumoraux, en particulier la chromogranine A. Ce peptide est exprimé par presque tous les néoplasmes neuroendocriniens, les taux sériques étant associés à la charge tumorale. Il faut garder à l'esprit que les niveaux de chromogranine A sont affectés par les inhibiteurs de la pompe à protons, une famille de médicaments couramment utilisés pour traiter la gastrite ou le reflux gastro-œsophagien (par exemple l'oméprazole, le pantoprazole). En outre, l'évolution des taux de chromogranine A sur une période donnée est utile pour évaluer la réponse aux thérapies. D'autres analyses sanguines peuvent porter sur le polypeptide pancréatique ou l'énolase spécifique aux neurones.
Comme nous l'avons vu dans les sections précédentes, la classification clinique et radiologique déterminera la prise en charge appropriée, qui diffère selon que la maladie est localisée, localement avancée ou métastatique.
Le traitement des tumeurs endocrines sporadiques
Les tumeurs endocrines localisées
Lorsque la maladie est localisée (sans implication des ganglions lymphatiques, infiltration artérielle ou métastases à distance), le pilier de la thérapie est la résection chirurgicale.
Il n'y a pas de consensus sur la meilleure stratégie chirurgicale pour les petites lésions, qui sont de plus en plus souvent détectées par l'imagerie en coupe transversale. Il semble y avoir une corrélation linéaire entre la taille de la tumeur et le comportement agressif, de sorte que des résections épargnant le parenchyme avec une lymphadénectomie limitée ou nulle ont été proposées pour les néoplasmes < 2 cm. En particulier, la pancréatectomie du segment moyen est indiquée pour enlever les tumeurs primaires situées dans le corps du pancréas, tandis que l'énucléation est techniquement possible lorsque le néoplasme primaire n'est pas trop profond dans le parenchyme, et suffisamment éloigné du canal pancréatique principal (afin de minimiser le risque de dommages peropératoires accidentels du canal). Si une énucléation est effectuée, il a été démontré que l'échographie peropératoire permet de confirmer une distance de sécurité entre la tumeur et le canal. Les résections épargnant le parenchyme sont associées à un taux accru de complications postopératoires telles que la fistule pancréatique, mais le résultat fonctionnel à long terme est excellent, le taux d'insuffisance exocrine et d'insuffisance endocrine étant presque nul.
Dans certaines études, cependant, la taille de la tumeur ne permettait pas de prédire le comportement biologique des néoplasmes non fonctionnels et des néoplasmes fonctionnels autres que l'insulinome. Une fraction des patients présentant de petits néoplasmes primaires (7%) a été trouvée avec une maladie nodale et a finalement développé des métastases. Par conséquent, la décision d'effectuer une opération épargnant le parenchyme en fonction de la taille de la tumeur a été remise en question, et un prélèvement régional des ganglions lymphatiques avec examen pathologique peropératoire a été proposé pour guider l'approche chirurgicale.
À l'opposé, de nombreux auteurs ont proposé une première approche non chirurgicale pour les petits (< 2 cm) néoplasmes neuroendocriniens pancréatiques non fonctionnels diagnostiqués de manière fortuite. L'avantage théorique de la prise en charge non chirurgicale serait d'éviter la morbidité (40-50%) et la mortalité (2%) associées aux résections pancréatiques chez les patients présentant un risque perçu très faible de malignité. Les patients pris en charge de manière non chirurgicale devraient être inscrits dans un protocole de surveillance clinique et radiologique très strict, et devraient être informés du risque de progression de la tumeur pendant le suivi. Ce risque semble se situer dans une fourchette de 6 à 8 %.
Les études d'observation à long terme font défaut, et la gestion des petits néoplasmes diagnostiqués de manière accidentelle doit être soigneusement décidée par une équipe multidisciplinaire d'experts et discutée avec le patient et sa famille. Le patient doit être conscient du risque et des avantages de chaque décision thérapeutique.
Dans les cas de méoplasmes primaires résécables > 2cm, ou dans les néoplasmes avec infiltration de la veine mésentérique supérieure/veine portale, les résections formelles (pancréaticoduodénectomie et pancréatectomie gauche) sont les procédures de choix.
Les tumeurs endocrines localement avancées
Les néoplasmes neuroendocriniens pancréatiques localement avancés se sont propagés aux organes adjacents ou aux vaisseaux péripancréatiques, mais pas aux organes distants. Lorsqu'il y a de bonnes chances que la tumeur puisse être complètement enlevée, une opération peut être entreprise. La résection avec élimination complète de la tumeur semble améliorer sensiblement la survie.
Les options chirurgicales comprennent la pancréaticoduodénectomie pour les néoplasmes situés dans la tête pancréatique, dans le duodénum et dans la région périampullaire ; et la pancréatectomie gauche avec splénectomie pour les lésions situées dans le corps et la queue du pancréas. Lorsque la section en extemporané peropératoire présente des marges de résection positives, la résection doit être étendue jusqu'à la pancréatectomie totale. Une lymphadénectomie régionale doit toujours être effectuée. L'infiltration des organes voisins (estomac, côlon, rein, surrénales) ne représente pas une contre-indication aux résections multiviscérales en bloc. Le rôle de la résectionvasculaire (veine mésentérique supérieure/veine portale) dans le cadre d'une pancréaticoduodénectomie, d'une pancréatectomie gauche ou d'une pancréatectomie totale a été établi, La réduction chirurgicale (résection avec tumeur résiduelle macroscopique, ou résection R2) ne semble pas améliorer la survie. De plus, le réduction est associé à un risque substantiel d'ensemencement de la tumeur péritonéale.
Chez des patients hautement sélectionnés présentant une maladie localement avancée, la thérapie par radionucléides à récepteurs de peptides (PPRT) a été utilisée pour réduire la tumeur. Cela signifie que le néoplasme a rétréci, ce qui rend la résection plus facile. La thérapie par radionucléides à récepteurs de peptides (PRRT) combine des analogues de la somatostatine (octréotide) avec un radionucléide pour former des molécules appelées analogues de la somatostatine radiomarqués. Ces radio-peptides sont injectés et se lient aux cellules néoplastiques neuroendocrines, qui possèdent des récepteurs pour eux. Une fois liés, les radio-peptides émettent des radiations et tuent les cellules tumorales auxquelles ils sont liés. Les deux radionucléides qui se fixent à l'octréotide pour créer les radiopeptides sont l'yttrium-90 (90Y-DOTA-TOC) et le lutétium-177 (177Lu-DOTA-TATE). Ce type de thérapie est surtout utilisé dans les néoplasmes neuroendocriniens métastatiques et ne peut être appliqué qu'aux personnes dont les tumeurs expriment des récepteurs à la somatostatine (comme le montre OCTREOSCAN™ ou 68-Ga-DOTA-TATE PET-CT). Le downstaging par PPRT est une procédure expérimentale, et doit être conseillé par une discussion multidisciplinaire expérimentée. Les résultats préliminaires semblent encourageants.
Le néoplasie endocrinienne multiple type 1 (syndrome MEN-1)
La néoplasie endocrinienne multiple de type 1 (syndrome MEN-1) ou syndrome de Wermer fait partie d'un groupe de troubles qui affectent le système endocrinien par le développement de lésions néoplastiques dans l'hypophyse, la glande parathyroïde et le pancréas. Bien qu'il soit généralement hérité d'un trouble autosomique dominant, le syndrome MEN1 peut également se manifester sporadiquement (sans antécédents familiaux) à la suite de nouvelles mutations. Jusqu'à 90 % de toutes les mutations identifiées prédisent une perte de fonction de la ménine, une protéine impliquée dans la prolifération cellulaire codée par un gène suppresseur de tumeur situé sur le chromosome 11. MEN1 suit le modèle de mutation "à deux coups". La première est la mutation hétérozygote de la lignée germinale MEN1 (familiale), ou développée à un stade embryonnaire précoce (sporadique) et présente dans toutes les cellules à la naissance. La seconde est une mutation somatique MEN1 qui se produit dans la cellule endocrine prédisposée et qui donne aux cellules l'avantage de survie nécessaire au développement de la tumeur. Le syndrome MEN1 est très pénétrant. Cinquante pour cent des patients développent des signes et des symptômes avant l'âge de 20 ans et plus de 95 % présentent des symptômes avant l'âge de 40 ans. Les tumeurs associées au syndrome MEN1 présentent la séquence adénome - carcinome caractéristique.
Les néoplasmes les plus courants sont :
- Hyperplasie parathyroïdienne et adénome >90 %.
- Néoplasmes endocriniens du pancréas = 80%.
- Néoplasmes hypophysaires = 25-50%.
- Autres néoplasmes = 5 à 30%.
Les néoplasmes neuroendocriniens du pancréas chez les hommes peuvent être synchrones ou métachrones avec d'autres néoplasmes associés aux hommes. Les néoplasmes pancréatiques sont généralement des gastrinomes, des insulinomes ou d'autres néoplasmes non fonctionnels. Les tumeurs sont très souvent multifocales et ont tendance à réapparaître. Ces aspects sont importants pour planifier la meilleure stratégie chirurgicale. La résection chirurgicale au moment du diagnostic est nécessaire dans les insulinomes et en présence d'un syndrome hormonal, tandis que le traitement des petits gastrinomes ou des néoplasmes non fonctionnels (<2 cm) est controversé.
Comme nous l'avons vu dans la section sur les néoplasmes sporadiques, les petits néoplasmes neuroendocriniens pancréatiques non fonctionnels découverts par hasard sont de plus en plus souvent diagnostiqués grâce à l'application généralisée de l'imagerie en coupe transversale, telle que la tomodensitométrie et l'imagerie par résonance magnétique. Des preuves récentes suggèrent que les petites lésions pancréatiques n'ont pas tendance à former des métastases, et que le développement de métastases n'affecte pas de manière critique la survie à long terme. En conséquence, une approche non chirurgicale (surveillance clinique et radiologique) est indiquée pour les lésions < 2cm, tandis qu'une résection chirurgicale doit être entreprise lorsque la tumeur se développe au cours du suivi, ou lorsque des métastases se développent. En particulier, la surveillance des petits gastrinomes associés au MEN1 est sûre, et le syndrome hormonal (Zollinger-Ellison) est bien contrôlé par la thérapie médicale. La résection chirurgicale n'est recommandée que pour les gastrinomes > 2cm.
La meilleure approche chirurgicale pour les néoplasmes neuroendocriniens pancréatiques associés au MEN1 n'est pas bien établie, et le choix d'une résection épargnant le parenchyme ou d'une résection formelle doit être adapté à chaque patient. La pancréatectomie totale doit être pratiquée chez les patients présentant des lésions mutifocales et des antécédents familiaux de maladie agressive. Les résections formelles (pancréaticoduodénectomie et pancréatectomie gauche) sont indiquées dans les grands néoplasmes, dans les néoplasmes avec atteinte ganglionnaire et dans les lésions multifocales centrées sur la tête ou la queue du pancréas. L'échographie peropératoire est obligatoire pour éliminer les lésions non détectées dans le reste du pancréas. L'énucléation est surtout pratiquée dans les insulinomes.
1. La pancréaticoduodénectomie céphalique
La pancréaticoduodénectomie céphalique (DPC) est le plus souvent pratiquée pour une malignité de la tête du pancréas et du périampullaire, mais peut également être indiquée dans certains cas de pancréatite chronique ou de tumeurs périampullaires bénignes. Il s'agit d'une opération majeure qui implique l'ablation de la tête du pancréas, du duodénum, de la vésicule biliaire et du canal cholédoque (figure ). Une courte longueur d'intestin grêle au-delà du duodénum est également retirée. Dans l'opération classique de Kausch-Whipple, le pylore (sortie de l'estomac) et la partie distale (inférieure) de l'estomac sont enlevés, tandis que dans l'opération de Longmire-Traverso (pancréatico-duodénectomie conservant le pylore), l'estomac et le pylore ne sont pas enlevés.
Après la résection, l'extrémité du canal biliaire restant ; le pancréas et l'estomac restants sont alors reliés à l'intestin grêle pour assurer l'écoulement de la bile, des sucs digestifs et des aliments dans les intestins . Trois anastomoses sont construites :
L'anastomose pancréatique. Le reste du pancréas est anastomosé au jéjunum (pancréatico-jéjunostomie) ou à la paroi postérieure de l'estomac (pancréatico-gastrostomie).
Anastomose biliaire. L'hépatico-jéjunostomie est pratiquée entre un reste de canal hépatique commun et un site du jéjunum distal par rapport à l'anastomose du pancréatico-jéjunal.
Anastomose entérique. Dans la DPC de Whipple, une anastomose antécolique est construite entre l'estomac et le jéjunum ; dans la DPC de Longmire-Traverso, une anastomose duodéno-jéjunale antécolique est créée.
Différentes modifications techniques des techniques de reconstruction ont été proposées, mais aucune n'a donné de résultats supérieurs dans les méta-analyses. Le choix de la technique de reconstruction à adopter dépend de la préférence du chirurgien et des pratiques institutionnelles.
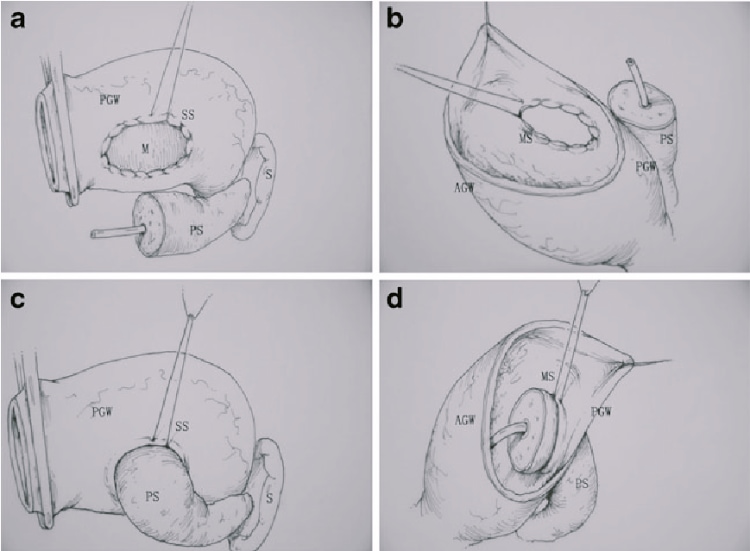
2. La pancréatectomie gauche (distale)
La pancréatectomie gauche (ou distale) est pratiquée pour traiter les maladies du pancréas de la queue et du corps. Cette opération implique une résection chirurgicale du corps et de la queue du pancréas à gauche de la confluence supérieure de la veine mésentéro-portée. La pancréatectomie gauche peut être réalisée avec ou sans splénectomie associée. Le choix de la procédure dépend du processus pathologique et des caractéristiques de la lésion.
Pancréatectomie gauche avec splénectomie : la rate, qui est située près de cette partie du pancréas et partage certains des mêmes vaisseaux sanguins, doit être retirée dans le cadre de la procédure lorsque le néoplasme pancréatique sous-jacent est agressif. La pancréatectomie gauche avec splénectomie permet de ligaturer les vaisseaux spléniques à leur origine et d'obtenir une clairance adéquate des ganglions lymphatiques.
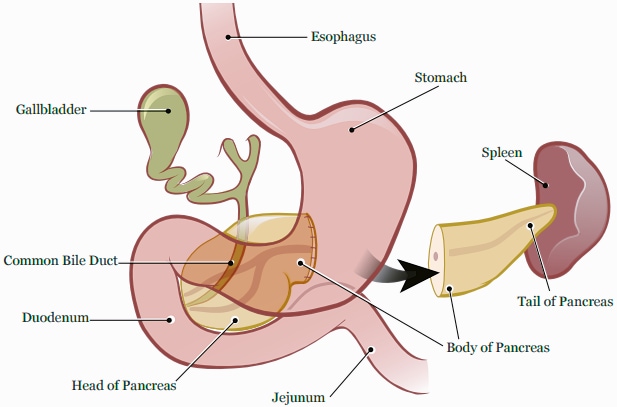
Pancréatectomie gauche avec préservation de la rate : cette procédure est réservée aux lésions et kystes pancréatiques bénins ou limites, ainsi qu'aux tumeurs neuroendocrines localisées. Il existe deux approches distinctes pour préserver la rate lors de la dissection du pancréas distal. La méthode classique consiste à identifier, isoler et préserver l'artère et la veine spléniques (procédure de Kimura). L'artère et la veine splénique peuvent également être ligaturées avec le pancréas, et la perfusion de la rate est assurée par les courts vaisseaux gastriques (procédure de Warshaw). Ces deux techniques sont acceptées comme étant appropriées pour traiter une masse dans la queue du pancréas.
Le moignon pancréatique peut être soit suturé à la main, soit fermé à l'aide d'une agrafeuse, soit scellé à l'aide d'un scalpel harmonique. Ces techniques de fermeture du moignon semblent être équivalentes.
La chirurgie mini-invasive est en train de devenir le paradigme des résections du pancréas gauche (pancréatectomie gauche laparoscopique et robotisée). Dans un avenir proche, le nombre de résections du pancréas gauche réalisées avec des techniques peu invasives devrait augmenter en raison de divers facteurs, notamment l'expérience accrue des opérateurs dans les différents centres et l'acceptation de la technique sur la base des résultats démontrés dans les lésions prémalignes et malignes.
3. La pancréatectomie Totale
La pancréatectomie totale implique la résection du pancréas entier, du canal cholédoque, de la vésicule biliaire, du duodénum, d'un court segment de l'intestin grêle au-delà du duodénum, du pylore (sortie de l'estomac), de la partie distale (inférieure) de l'estomac, de la rate et des ganglions lymphatiques régionaux (figure ).
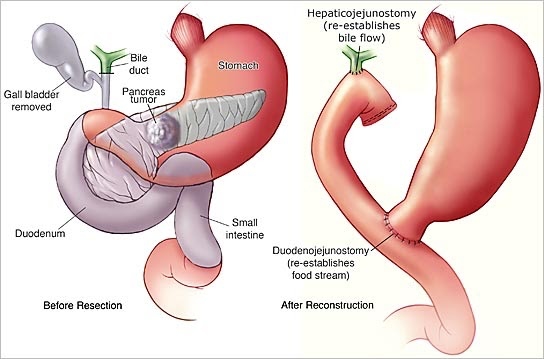
Après la résection, l'extrémité du canal biliaire restant et l'estomac sont reliés à l'intestin grêle pour assurer l'écoulement de la bile et des aliments dans les intestins . Deux anastomoses sont construites :
L'anastomose biliaire. L'hépatico-jéjunostomie est pratiquée entre le reste du canal hépatique commun et un site sur le jéjunum.
Anastomose entérique. Une anastomose antécolique est construite entre l'estomac et le jéjunum ; distale par rapport à l'hépatico-jéjunostomie.
Les indications d'une pancréatectomie totale élective en une étape comprennent la présence d'un néoplasme multifocal (néoplasie mucineuse papillaire intraductale, métastases pancréatiques provenant d'un carcinome rénal à cellules claires, tumeur neuroendocrine dans le cadre de MEN1), ou la présence d'un néoplasme mucineux papillaire intraductal impliquant l'ensemble du canal pancréatique principal. Une pancréatectomie totale non planifiée en une seule étape peut être nécessaire après une pancréatectomie partielle initiale en raison des marges de résection positives sur la section congelée peropératoire. La pancréatectomie totale en deux temps (pancréatectomie d'achèvement) est pratiquée en raison de complications postopératoires graves ou de récidive néoplasique dans le reste du pancréas après une résection pancréatique antérieure.
La pancréatectomie totale est invariablement associée au développement d'une insuffisance exocrine (incapacité à digérer correctement les aliments) et d'une insuffisance endocrine (diabète sucré). La gestion de l'insuffisance pancréatique comprend la thérapie de remplacement des enzymes pancréatiques et l'insulinothérapie. L'insuffisance exocrine et le diabète peuvent être particulièrement difficiles à contrôler dans les premiers mois suivant l'opération, mais des études ont indiqué que la qualité de vie à long terme est satisfaisante.
4. La pancréatectomie Centrale
La pancréatectomie centrale consiste en une résection limitée de la partie médiane du pancréas. Cette procédure permet au chirurgien de préserver le parenchyme pancréatique et par conséquent la fonction pancréatique endocrine et exocrine à long terme. Les indications de la pancréatectomie cenrtale comprennent les néoplasmes bénins et limites du corps pancréatique.
Après identification et isolement des principales structures vasculaires autour du corps et du cou du pancréas, le segment du pancréas comportant la tumeur est sectionné à gauche et à droite de la lésion (figure ). Le moignon céphalique est suturé avec des points de suture interrompus après une ligature élective du canal de Wirsung ou au moyen d'une agrafeuse.
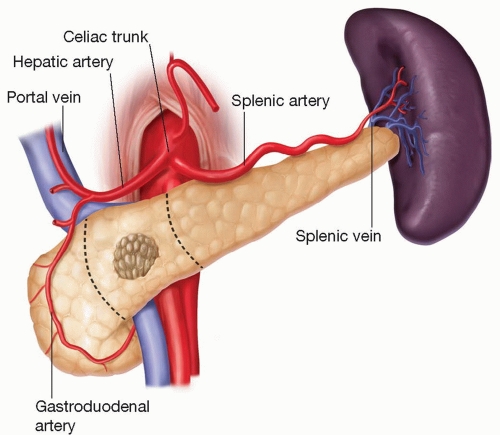
La phase de reconstruction implique une ou deux anastomoses :
Anastomose pancréatique. Le reste du pancréas distal est anastomosé à une boucle de Roux-en-Y jéjunale (pancréatico-jéjunostomie) ou à la paroi postérieure de l'estomac (pancréatico-gastrostomie).
Anastomose entérique. Après une pancréatico-jéjunostomie, l'anse de Roux est reliée au jéjunum distal.
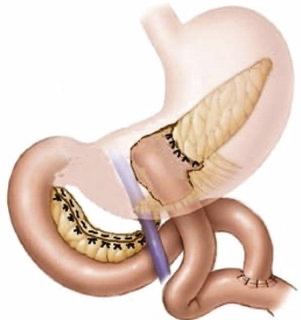
5. Les Enucléations pancréatiques
Une procédure d'énucléation pancréatique est une opération visant à enlever les petites tumeurs bénignes (<2 cm) du pancréas. Cette procédure consiste à enlever la tumeur du pancréas environnant. Pour que l'énucléation soit effectuée en toute sécurité, la lésion doit se trouver à au moins 2 à 3 mm du canal pancréatique principal et pas trop profondément dans le parenchyme. Si la lésion est trop proche d'un canal, le risque de lésion canalaire accidentelle est important et peut entraîner une fistule pancréatique particulièrement difficile à traiter. Par conséquent, la distance entre la tumeur et le canal pancréatique principal doit être évaluée en préopératoire au moyen de l'imagerie par résonance magnétique avec cholangio-pancréatographie. Une échographie peropératoire doit également être réalisée pour confirmer la relation entre le néoplasme et le pancréas principal. De plus, elle permet d'identifier clairement la lésion et d'évaluer sa morphologie et son emplacement.
L'incidence des complications postopératoires, en particulier la fistule pancréatique, est élevée, malgré le fait que la majorité des fistules ont un cours indolent. Le principal avantage de l'énucléation est la préservation de presque tout le parenchyme pancréatique, ce qui minimise le risque d'insuffisance exocrine et endocrine à long terme.
L'énucléation peut être réalisée en utilisant une approche peu invasive.
6. Les résections pancréatiques mini-invasive (laparoscopie 4k-3D et Robotic)
La chirurgie mini-invasive est pratiquée par de petites incisions, généralement entre 5 et 15 mm. Le chirurgien insère ensuite des instruments fins spécialement conçus et un équipement vidéo sophistiqué pour réaliser l'opération à travers la plus petite ouverture. Selon la procédure, la chirurgie mini-invasive peut être effectuée avec le chirurgien manipulant les instruments à la main (chirurgie laparoscopique) ou avec le chirurgien dirigeant des bras robotisés (chirurgie assistée par robot).
La chirurgie mini-invasive présente de nombreux avantages par rapport aux techniques traditionnelles, notamment une moindre lésion des tissus, moins de douleurs postopératoires, des séjours hospitaliers plus courts, un retour plus rapide aux activités normales, des cicatrices minimes, moins de hernies incisionnelles. Au cours des 20 dernières années, la chirurgie mini-invasive a évolué à un point tel que la majorité des interventions de chirurgie générale peuvent être réalisées en toute sécurité dans des mains dûment qualifiées.
La complexité de la chirurgie du pancréas a fait que le développement des techniques laparoscopiques et en particulier des résections formelles a été relativement lent par rapport aux procédures de nombreuses autres spécialités chirurgicales. Par conséquent, son intégration dans la pratique clinique régulière est récente, et les premières indications étaient limitées aux néoplasmes bénins et limites. Des données récentes indiquent que les résections pancréatiques par laparoscopie sont oncologiquement adéquates et peuvent être appliquées en toute sécurité aux néoplasmes malins. Les résections pancréatiques laparoscopiques les plus couramment pratiquées sont la pancréatectomie gauche et l'énucléation, tandis que la pancréaticoduodénectomie est pratqué régulièrement que dans un nombre limités d’hôpitaux dans le monde dont le Centre Hospitalier du Luxembourg.
La chirurgie mini-invasive a été encore améliorée par l'utilisation de la robotique. La chirurgie robotique est une technique dans laquelle un chirurgien effectue une intervention chirurgicale à l'aide d'un ordinateur qui contrôle à distance de très petits instruments fixés à un robot. Le chirurgien étant assis à une console à quelques mètres du patient, le robot traduit les mouvements de la main du chirurgien en micro-mouvements correspondants des instruments à l'intérieur du corps du patient. Le système robotique offre une meilleure visualisation, dextérité, précision et contrôle que la chirurgie ouverte, tout en permettant au chirurgien d'effectuer des procédures par de minuscules incisions de 1 à 2 cm. L'expérience mondiale en matière d'utilisation de systèmes robotiques pour la chirurgie du pancréas s'accroît.
L'unité de Chirurgie du Centre Hopsitalier du Luxembourg possède l'une des plus grandes pratiques mini-invasives d'Europe. Notre programme a été le pionnier d'approches innovantes et peu invasives telles que la pancréatectomie gauche avec splénectomie, la pancréatectomie gauche préservant la rate, l'énucléation, la pancréatectomie moyenne et la pancréaticoduodénectomie. Nous disposons des tous les plus modernes technologie pour la laparoscopie, telle la vione 4K et 3D et le dernier modele du robot Da Vinci.
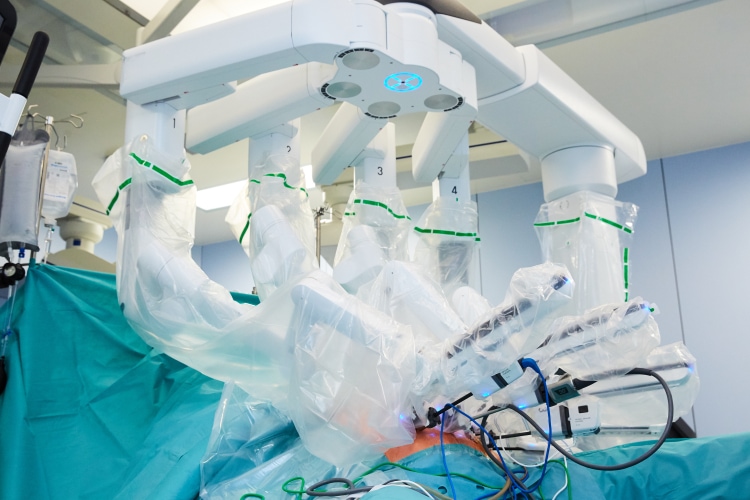
7. Les résections vasculaires
Résections du pancréas associées à des résections vasculaires : environ la moitié des tumeurs du pancréas (tumeurs borderline et localement avancées) nécessitent la résection d'un vaisseau péri-pancréatique (veine porte, veine mésentérique, plus rarement artère hépatique, artère mésentérique supérieure) pour pouvoir retires le tumeur. Ces interventions très complexes ne sont possibles que dans un groupe limité de patients ayant déjà reçu un traitement préopératoire (chimiothérapie néo-adjuvante et/ou radiothérapie néo-adjuvante). Au Centre Hopsitalier du Luxembourg, des duodénocéphalopancréasectomies avec résection vasculaire et des splénopancréasectomies avec résection vasculaire sont pratiquées régulièrement par un team avec un grande expérience dans ce demain complexe de la chirurgie.
Malgré les récents progrès techniques, les complications postopératoires après une chirurgie du pancréas (en particulier après une résection du pancréas) restent un problème majeur. Le principal déterminant de la morbidité postopératoire est - paradoxalement - le pancréas, un organe très mou et friable qui produit des enzymes digestives. Un diagnostic précoce et la gestion correcte des complications postopératoires nécessitent une grande expérience dans ce domaine et expliquent un résultat postopératoire favorable.
- La Fistule Pancréatique
- L’hémorragie post-résection du pancréas
- Le retard de la vidange gastrique
- Autres complications: La pancréatite post-opératoire / La fistule biliaire / Les complications pulmonaires
- L’insuffisance pancréatique exocrine
- L’insuffisance pancréatique endocrine
1. La Fistule Pancréatique
La fistule pancréatique est la principale complication après les résections pancréatiques. Elle est définie comme la sortie, via un drain, une voie de drainage ou une plaie chirurgicale (le ou après le troisième jour postopératoire), de tout volume mesurable de liquide contenant du jus pancréatique (teneur en amylase supérieure à 3 fois la valeur sérique normale supérieure). Le liquide de drainage peut avoir une "apparence sinistre" qui peut varier d'un liquide brun foncé à verdâtre, d'une eau laiteuse à une "eau de source" claire qui ressemble à du jus pancréatique. Les résultats cliniques associés peuvent inclure des douleurs abdominales et une distension avec une fonction intestinale altérée, un retard de la vidange gastrique, de la fièvre, un taux élevé de leucocytes dans le sérum et une augmentation des protéines C-réactives.
L'origine de la fistule pancréatique diffère selon le type de résection pancréatique :
Dans la pancréaticoduodénectomie, la fistule pancréatique représente l'échec de la guérison/la fermeture de la pancréatico-jéjunostomie ou de la pancréatico-gastrostomie. L'incidence se situe entre 10 et 20 %.
Dans la pancréatectomie gauche, la fistule pancréatique représente une fuite parenchymateuse qui n'est pas directement liée à une anastomose. La fuite provient de la surface brute du pancréas (fermée avec des sutures ou avec une agrafeuse). L'incidence est de 20 à 25 %.
Dans le segment moyen de la pancréatectomie, la fistule pancréatique provient d'un échec de la cicatrisation de l'anastomose pancréatico-entérique réalisée sur le moignon distal, et/ou elle peut représenter une fuite parenchymateuse provenant de la surface pancréatique brute du moignon proximal. L'incidence est de 40 à 50 %.
Dans l'énucléation, la fistule pancréatique représente une fuite parenchymateuse à partir du lit de résection, souvent en raison d'une lésion accidentelle d'un conduit secondaire ou, plus rarement, en raison d'une lésion du conduit pancréatique principal. Son incidence est de 35 à 40 %.
Les facteurs de risque de fistule pancréatique les plus largement reconnus sont directement liés à l'état et à la maladie du pancréas et/ou de la région périamullaire. Le principal d'entre eux est un parenchyme pancréatique mou. La texture normale du pancréas est molle et friable, et reste intacte lorsque des néoplasmes petits et bénins/bords se développent. D'autre part, l'adénocarcinome canalaire et - surtout - la pancréatite chronique provoquent l'obstruction des canaux et le remplacement fibrotique du tissu pancréatique normal. Il est largement admis qu'un reste pancréatique fibrotique facilite l'anastomose pancréatico-entérique, tandis qu'un parenchyme pancréatique mou et friable rend l'anastomose difficile à réaliser et sujette à des lésions inflammatoires (en raison de la composante acineuse inaltérée), et à des fuites.
La taille du canal pancréatique principal a également été impliquée comme un prédicteur majeur de fistule. Les petits conduits pancréatiques non dilatés, généralement définis comme ayant un diamètre inférieur ou égal à 3 mm, prédisposent les patients aux fistules pancréatiques. Un petit canal pancréatique et un parenchyme mou coexistent souvent, ce qui augmente le risque de fistule postopératoire.
Malgré les améliorations apportées aux techniques chirurgicales et aux soins postopératoires, la fistule pancréatique reste un problème majeur. Les variations de la technique chirurgicale (modification de la technique d'anastomose pancréatique dans la pancréaticoduodénectomie ou gestion du moignon pancréatique dans la pancréatectomie distale) ou de la gestion postopératoire n'ont pas modifié l'incidence de la fistule de manière appréciable.
Cliniquement, la fistule pancréatique peut être associée à des collections abdominales, des abcès, une infection, de la fièvre, une distension abdominale et une déficience intestinale, des saignements. Un système de classement clinique des fistules pancréatiques postopératoires (A, B, C) a été proposé par le Groupe international d'étude des fistules pancréatiques (ISGPF) et est résumé ci-dessous :
La fistule pancréatique de grade A n'a pas d'impact clinique et nécessite peu de changement dans la gestion ou de déviation par rapport à la voie clinique normale.
La fistule pancréatique de grade B nécessite un changement de gestion ou un ajustement du cheminement clinique. Souvent, le patient est soutenu par une nutrition artificielle. Les drains péripancréatiques sont généralement maintenus en place ou repositionnés. Des antibiotiques sont généralement nécessaires, des analogues de la somatostatine peuvent également être utilisés. Une fistule de grade B entraîne généralement un retard dans la sortie, ou une réadmission après une sortie précédente peut être nécessaire.
Une fistule pancréatique de grade C nécessite un changement majeur dans la gestion clinique. Une procédure invasive est nécessaire, y compris la mise en place d'un drain percutané ou une réexploration chirurgicale. La patiente doit généralement faire l'objet d'un séjour prolongé à l'hôpital, avec un retard important dans la sortie de l'hôpital. Il y a souvent des complications associées et la possibilité d'une mortalité post-opératoire.
De nombreux patients souffrant de fistules pancréatiques postopératoires de grade B et C peuvent recevoir leur congé avec des drains in situ et être observés en ambulatoire.
2. L’hémorragie post-résection du pancréas
L'hémorragie postopératoire est l'une des complications les plus graves après les résections pancréatiques, avec une incidence comprise entre 2 et 8 %. Le groupe d'étude international de la chirurgie pancréatique (ISGPS) a élaboré une définition objective et généralement applicable de l'HPP, basée sur 3 paramètres :
- Début (précoce/tardif)
- Lieu (intraluminal/extraluminal)
- Gravité (légère/grave)
L'hémorragie postopératoire se manifeste par une perte de sang au niveau des drains ou de la sonde nasogastrique, et/ou par des signes cliniques d'hypovolémie (perte de volume sanguin). À moins que l'hémorragie ne nécessite un traitement d'urgence, tous les patients doivent être surveillés au départ (hémoglobine, numération des globules rouges, hématocrite, tension artérielle, pouls, débit urinaire). Un CT-scan peut permettre de mettre en évidence le site de l'hémorragie et les collections abdominales associées.
Une hémorragie post-pancréatectomie précoce se produit dans les 24 premières heures suivant l'opération. Elle est très probablement due à une défaillance technique de l'hémostase appropriée pendant l'opération de référence ou à une coagulopathie périopératoire sous-jacente. Si cela est possible, elle peut être traitée par des transfusions sanguines ; sinon, une réexploration et une hémostase sont nécessaires. Une réexploration dans les 24 heures suivant l'opération de référence ne modifie pas sensiblement le déroulement postopératoire.
Les hémorragies post-pancréatectomie tardives sont généralement dues à des complications de l'opération, avec un retard habituel de plusieurs jours, voire de plusieurs semaines (par exemple, après des abcès intra-abdominaux, l'érosion d'un vaisseau péri-pancréatique secondaire à une fistule pancréatique ou à des drains intra-abdominaux, une ulcération au site d'une anastomose, ou en association avec un pseudo-anévrisme artériel qui s'est développé). Un "saignement sentinelle", une petite perte de sang par les drains abdominaux ou la sonde nasogastrique plusieurs heures avant une hémorragie massive, peut être présent (30 à 100%). Une angiographie diagnostique peut localiser le site de l'hémorragie et une embolisation peut être effectuée. Sinon, une réexploration chirurgicale peut être nécessaire, notamment pour traiter une hémorragie massive ou une complication concomitante (par exemple, une fistule pancréatique). L'endoscopie peut jouer un rôle dans les saignements intraluminaux. L'hémorragie tardive est un événement grave associé à un taux de mortalité de 15 à 20 %.
Les saignements intraluminaux se produisent dans la lumière intestinale. Lors d'une pancréaticoduodénectomie et d'une pancréatectomie du segment moyen, les saignements peuvent provoquer des lignes de suture de l'anastomose pancréato-entérique en raison de la digestion enzymatique de la paroi des vaisseaux sanguins à la surface du moignon pancréatique par des enzymes exocrines pancréatiques secondaires à une fuite pancréatique. Les autres sites de saignement intraluminal sont l'anastomose gastro-entérique ou l'anastomose duodéno-entérique. Ces saignements dépendent généralement d'un ulcère gastrique ou duodénal ou d'une gastrite diffuse. Les saignements intraluminaux se manifestent par du sang sombre dans la sonde naso-gastrique (s'il y a lieu), ou par une hématémèse et/ou une méléna (selles noires), et/ou des signes de choc hypovolémique. Le traitement des saignements de l'anastomose entérique est endoscopique, tandis que les saignements de l'anastomose pancréatique peuvent nécessiter une réexploration chirurgicale.
Les saignements extraluminaux se produisent dans la cavité abdominale et peuvent provenir de vaisseaux artériels ou veineux dans les zones de résection (en particulier les lamelles rétro-portées), ou de pseudo-anévrismes érodés et rompus, qui sont secondaires à une infection intra-abdominale avec implication des vaisseaux péri-pancréatiques, ou à une lésion vasculaire pendant la résection. Les hémorragies intraluminales présentent une perte de sang par les drains intra-abdominaux et/ou des signes de choc hypovolémique. Le traitement peut être chirurgical ou angiographique.
La gravité des saignements peut être différenciée en 2 catégories selon l'importance de la perte de sang ou les besoins de transfusion : les saignements légers n'entraînent pas d'atteinte clinique, la baisse du taux d'hémoglobine de <3 g/dl, et la transfusion de <3 unités de globules rouges concentrés dans les 24 heures. Les saignements graves impliquent une perte de sang importante (baisse du taux d'hémoglobine de >3 g/dl), une altération cliniquement significative (par exemple, tachycardie, hypotension, oligurie, choc hypovolémique), la nécessité d'une transfusion sanguine de >3 unités de globules rouges dans les 24 heures, et la nécessité d'un traitement invasif (embolisation angiographique interventionnelle ou relaparotomie).
L'hémorragie post-pancréatectomie reste une complication majeure qui nécessite au moins un suivi clinique attentif. Reconnaître cet événement à temps peut permettre d'éviter des conséquences graves et fatales. Une équipe d'experts multidisciplinaire est obligatoire pour assurer le meilleur traitement 24 heures sur 24.
3. Le retard de la vidange gastrique
Le retard de vidange gastrique consiste en une gastroparésie fonctionnelle, et c'est l'une des complications les plus fréquentes après une résection pancréatique (surtout après une pancréaticoduodénectomie). Son incidence varie considérablement selon les établissements chirurgicaux (5 à 25 %). La définition du retard de vidange gastrique englobe différents éléments cliniques, dont
- Prolongation de la sonde naso-gastrique ou nécessité de la réinsertion de la sonde naso-gastrique
- Incapacité à tolérer une alimentation solide
- Vomissements et distension gastrique
- Utilisation de médicaments procinétiques
Les causes du retard de la vidange gastrique ne sont toujours pas claires et sont probablement multifactorielles, impliquant une perturbation de l'innervation du pylore, une carence en motiline due à la résection du duodénum et des aspects techniques. Plusieurs études ont suggéré une plus grande incidence de vidange gastrique retardée après une pancréaticoduodénectomie de Whipple avec préservation du pylore, alors que d'autres ont trouvé l'effet inverse. La méthode de reconstruction du drainage gastrique (antecolique ou rétrocolique) est un autre facteur opératoire qui peut influer sur le taux de vidange gastrique retardée. Enfin, le retard de vidange gastrique est souvent secondaire à une fistule pancréatique, une fistule biliaire et des collections abdominales.
Lorsque l'on soupçonne un retard de vidange gastrique, il peut être nécessaire d'exclure une obstruction de la sortie gastrique. Cela peut se faire par une radiographie de la déglutition de la gastrograveine (qui permet au radiologue de voir le mouvement du colorant à travers l'anastomose digestive), ou par une endoscopie supérieure (qui permet de visualiser l'anastomose digestive et les problèmes associés, tels que les ulcères anastomotiques).
Trois grades différents (A, B et C) ont été définis en fonction de l'impact sur l'évolution clinique et sur la prise en charge postopératoire :
Un retard de vidange gastrique de grade A n'entraîne généralement pas de changement marqué de la prise en charge, si ce n'est des perturbations mineures dans le retour à la prise d'aliments solides ou la réinsertion de la sonde nasogastrique pendant une brève période.
Le retard de vidange gastrique de grade B nécessite un traitement avec des médicaments prokinétiques et un soutien nutritionnel parentéral ou entéral, et généralement une réinsertion prolongée de la sonde nasogastrique.
Le retard de vidange gastrique de grade C nécessite un changement majeur dans la prise en charge clinique, et éventuellement le traitement des complications postopératoires associées, telles que la fistule pancréatique ou les abcès intra-abdominaux. Par conséquent, un bilan diagnostique plus approfondi et des interventions radiologiques ou chirurgicales sont souvent nécessaires.
Un retard de la vidange gastrique peut entraîner une gêne importante pour le patient, et son traitement peut être difficile. Une équipe multidisciplinaire comprenant des gastroentérologues et des diététiciens expérimentés est nécessaire pour fournir les meilleurs soins.
4. Autres complications
La pancréatite post-opératoire
La pancréatite postopératoire dépend d'une inflammation locale déclenchée par la digestion enzymatique au niveau du reste du pancréas. Les patients ayant un pancréas mou et acineux sont plus susceptibles de développer une pancréatite postopératoire, malgré le fait que d'autres facteurs liés au patient peuvent contribuer à la pathogénèse. Une pancréatite postopératoire précoce (jour 1-3) contribue généralement à la formation d'une fuite pancréatique. Le diagnostic repose sur des taux élevés d'amylase sérique, un liquide de drainage brun foncé et une douleur postopératoire mal contrôlée. La pancréatite postopératoire nécessite des modifications de la prise en charge clinique, y compris une nutrition artificielle.
La fistule biliaire
La fistule biliaire est définie comme l'écoulement par un drain de tout volume mesurable de liquide contenant de la bile. Le liquide de drainage est généralement verdâtre et épais. La plupart du temps, la fistule biliaire ne provoque pas de manifestations cliniques graves, bien que dans quelques cas les résultats cliniques associés puissent inclure des douleurs abdominales et une distension avec une fonction intestinale altérée, un retard de vidange gastrique, de la fièvre, un nombre élevé de leucocytes sériques et une augmentation de la protéine C-réactive. Des fuites de bile peuvent se produire après une pancréaticoduodénectomie ou une pancréatectomie totale, et représentent l'échec de la guérison/la fermeture de l'hépatite-jéjunostomie. Occasionnellement, la fistule biliaire peut dépendre d'une fuite du reste du canal cystique. L'incidence de la fistule biliaire est de 3 à 5 %. Les options de traitement comprennent le jeûne et la nutrition artificielle pour permettre la guérison de l'anastomose. Il peut être nécessaire de mettre en place un drainage biliaire transhépatique percutané si les fistules à haut débit ne se résolvent pas spontanément.
Les complications pulmonaires
Les patients qui subissent une chirurgie du pancréas courent un risque accru de complications pulmonaires après l'opération. Une chirurgie abdominale supérieure majeure modifie la fonction pulmonaire postopératoire et réduit l'efficacité des efforts pour tousser pendant une semaine. Ces mécanismes entraînent une diminution de la capacité fonctionnelle résiduelle et vitale pendant de nombreux jours, puis une atélectasie ou une pneumonie. Les patients fragiles, âgés ou présentant des comorbidités courent un risque accru de développer une pneumonie. Les complications pulmonaires postopératoires augmentent la morbidité hospitalière et prolongent le séjour à l'hôpital. C'est pourquoi une physiothérapie thoracique postopératoire a été mise en place. Il a été démontré que les exercices respiratoires pendant l'hospitalisation améliorent les performances respiratoires et préviennent les complications pulmonaires postopératoires.
5. L’insuffisance pancréatique exocrine
L'insuffisance pancréatique exocrine avec maldigestion est causée par une déficience de la production d'enzymes pancréatiques due au remplacement fibreux du tissu pancréatique normal ou à la perte du parenchyme pancréatique. Cette affection est une conséquence majeure de la pancréatite chronique, mais peut également faire suite à des résections pancréatiques. Dans les résections pancréatiques, le développement d'une insuffisance exocrine dépend du type et de l'extension de la résection, de la résection intestinale associée et de facteurs individuels.
La digestion est souvent altérée après une pancréaticoduodénectomie, et les patients peuvent souffrir de stéatorrhée et de perte de poids. Cet état est souvent transitoire et dépend davantage de la résection intestinale que de la perte du parenchyme pancréatique. Le duodénum, en fait, fonctionne comme un stimulateur intestinal et active les enzymes pancréatiques. La plupart des patients retrouvent une fonction intestinale et pancréatique normale en quelques mois. L'insuffisance exocrine à long terme peut dépendre d'une défaillance de l'anastomose avec obstruction ultérieure du canal pancréatique et pancréatite chronique secondaire. Les signes d'insuffisance exocrine ne sont présents que lorsque 85 à 90 % du pancréas est incapable de sécréter des enzymes. Après une pancréatectomie gauche, l'insuffisance exocrine est rare.
La pancréatectomie totale est invariablement associée à une insuffisance exocrine et à une altération de la motilité gastro-intestinale. Cependant, ces conditions ont tendance à s'améliorer avec le temps (avec une thérapie de remplacement appropriée) de sorte que la qualité de vie rapportée à un an semble être comparable à celle des patients ayant subi une pancréaticoduodénectomie.
Les symptômes de l'insuffisance pancréatique exocrine sont principalement liés à la maldigestion des lipides et des protéines, et comprennent (à un degré variable) la malabsorption, la diarrhée, la stéatorrhée et la perte de poids. Des douleurs abdominales récurrentes peuvent être présentes. La dégradation des glucides peut être partiellement prise en charge par l'amylase dans la salive et les enzymes dans l'intestin grêle, tandis que la dégradation des protéines peut être partiellement prise en charge par la peptidase gastrique et l'entéropeptidase. La carence en vitamines liposolubles est généralement présente.
La quantification de l'insuffisance pancréatique exocrine nécessite des tests fonctionnels. Parmi ceux-ci, la détermination de l'élastase-1 fécale, de la chymotripsine fécale et de la teneur en graisses des selles sont les plus largement disponibles.
Les piliers du traitement de l'insuffisance pancréatique exocrine sont un régime alimentaire équilibré à faible teneur en graisses, une supplémentation en oligo-éléments et en vitamines, et une thérapie enzymatique de substitution. Une préparation d'enzymes pancréatiques (pancréatine) contient des lipases, de l'alpha-amylase et des protéases. La force de la préparation est définie par la teneur en lipase par capsule (microsphères ou minimicrosphères), où 10.000 = 10.000 unités de lipase. Jusqu'à 40 000 unités de lipase par capsule sont disponibles dans le commerce.
Les capsules doivent être prises avec les repas ou immédiatement après. Les préparations enzymatiques sont résistantes aux acides et s'activent sous un pH basique, dans l'intestin grêle. Néanmoins, des inhibiteurs de la pompe à protons peuvent être associés pour maximiser leur activation, en particulier dans le cas d'une hyperacidité gastrique. Le succès thérapeutique est principalement déterminé sur une base clinique.
6. L’insuffisance pancréatique endocrine
Certains patients développent une insuffisance pancréatique endocrinienne après une résection pancréatique, consistant en un métabolisme des glucides altéré jusqu'au diabète sucré. Le diabète après une résection pancréatique dépend de la perte des îlots de Langerhans et de la déficience de la production d'insuline.
Après une résection partielle du pancréas, il est fréquent de constater une altération temporaire du métabolisme des glucides, en particulier chez les patients qui reçoivent une alimentation parentérale. Cette condition peut être résolue par un régime alimentaire adéquat et une légère thérapie antidiabétique. Si le diabète se développe, l'insuline est nécessaire pour contrôler l'hyperglycémie et prévenir les complications à long terme. L'insuline est administrée par voie sous-cutanée à l'aide de stylos préremplis. Chez certains patients, le diabète peut se développer plusieurs années après la résection, et dépend d'une pancréatite obstructive chronique du reste.
La relation entre les résections pancréatiques partielles et le diabète sucré n'est toujours pas claire. Les patients subissant une pancréatectomie gauche semblent avoir un risque plus élevé de développer une forme de diabète à long terme, car la majeure partie des îlots de Langerhans est naturellement située dans la queue du pancréas.
La pancréatectomie totale entraîne une perte complète de la fonction endocrinienne du pancréas, avec des manifestations complètes de diabète pancréatique. L'absence d'insuline et de glucagon (qui s'opposent l'un à l'autre) augmente le risque d'hyperglycémie et d'hypoglycémie. Des fluctuations glycémiques importantes peuvent se produire, avec une hyperglycémie post-prandiale marquée et une hypoglycémie soudaine après l'administration d'insuline ou pendant un jeûne prolongé. Il est donc important de prévoir un plan de traitement du diabète comprenant une surveillance fréquente de la glycémie et une insulinothérapie souple pour aider à réduire le risque d'hyperglycémie et d'hypoglycémie